Mecanismos de resistencia bacteriana a las quinolonas
R. Taléns-Visconti1,2, T.M. Garrigues2 y E. Cantón1
1Unidad de Bacteriología Experimental, Centro de Investigación, Hospital Universitario La Fe, Valencia; 2Departamento de Farmacia y Tecnología Farmacéutica, Facultad de Farmacia, Universidad de Valencia, Burjassot (Valencia)
|
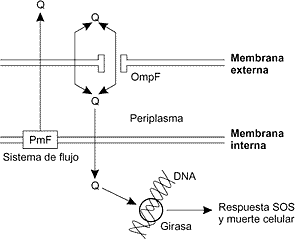
RESUMEN Para ejercer su efecto citotóxico las quinolonas deben penetrar a través de la membrana bacteriana y alcanzar su diana celular, la DNA girasa o la topoisomerasa IV, e inducir la muerte de la célula. Por ello, los mecanismos de resistencia a las fluoroquinolonas incluyen mutaciones en los genes que codifican la DNA girasa y la topoisomerasa IV dando lugar a las QRDR, alteraciones en la permeabilidad de la membrana que disminuyen la penetración intracelular del fármaco y actividad de transportadores activos endógenos que provocan la expulsión de los antimicrobianos desde la membrana celular al medio exterior. Estos mecanismos de resistencia pueden manifestarse solos o en combinación, si bien parece que in vivo el aumento en el grado de resistencia a las quinolonas es producto de varios mecanismos simultáneos. En el presente trabajo se revisan estos mecanismos de resistencia y se establece, en la medida de lo posible, una relación con la estructura de la quinolona. Palabras clave: Quinolona - Resistencias - Topoisomerasa IV - DNA girasa - QRDR - Penetración intracelular - Expulsión Mechanisms of bacterial resistance to quinolonesSUMMARY In order to produce their cytotoxic effect, quinolones must enter the cell through the bacterial membrane to reach their target, DNA-gyrase or topoisomerase IV, and induce cell death. The mechanisms of resistance to fluoroquinolones include: those mediated by gene mutations codifying for DNA gyrase and topoisomerase IV and leading to QRDR; those characterized by changes in the permeability of the outer membrane which decrease intracellular penetration of the drug; and those caused by active endogenous carriers responsible for drug efflux. These resistance mechanisms can occur alone or in combination; in fact, it is believed that high resistance levels to quinolones in vivo are associated with simultaneous mechanisms. This article reviews such resistance mechanisms and establishes, when possible, their relation to the structure of quinolones. Key words: Quinolones - Resistance - Topoisomerase IV - DNA gyrase - QRDR - Intracellular penetration - Efflux Las bacterias resistentes a las quinolonas aparecen en clínica como resultado de la terapia con estos agentes. Su efecto citotóxico depende de que penetren a través de la membrana bacteriana y alcancen su diana celular (DNA girasa o topoisomerasa IV) para inducir la muerte de la célula. En principio, las resistencias a las quinolonas pueden deberse a mutaciones que afecten cualquier paso de este proceso. Así, los mecanismos de resistencia bacteriana a las quinolonas pueden agruparse en tres categorías: 1) Resistencias de tipo cromosómico que dan lugar a mutaciones en segmentos definidos de los genes que codifican la DNA girasa (especialmente en la subunidad A) y la topoisomerasa IV, dando lugar a las QRDR (del inglés "Quinolone Resistance-Determining Region") (1, 2). 2) Resistencias por alteraciones en la membrana externa bacteriana que disminuyen la penetración intracelular del fármaco. Estas modificaciones se originan en alteraciones de los genes que codifican los canales de las porinas, lo que impide la entrada del quimioterápico en la bacteria. 3) Resistencias basadas en la expulsión del antibacteriano desde el medio intracelular al extracelular por acción de transportadores endógenos activos. Sin embargo, no se han descrito enzimas bacterianas capaces de degradar o inactivar a las quinolonas en el medio intracelular (2). En relación con el primer mecanismo mencionado, los estudios iniciales, centrados en Escherichia coli, demostraron que la resistencia a las quinolonas se produce normalmente por mutaciones en regiones definidas de las proteínas GyrA o GyrB (2-4). Las mutaciones en las regiones equivalentes de las proteínas ParC o ParE tienen lugar tras las acontecidas en la girasa, y producen, en general, altos grados de resistencia a estos fármacos (5). Por ello se identificó la girasa como principal diana de las quinolonas. El dominio rotura-reunión de la proteína GyrA de E. coli (GyrA59K) cristaliza como dímero, y es el mínimo fragmento de la subunidad A que al unirse con la subunidad B presenta actividad "DNA-cleavage". Cada monómero GyrA está compuesto por una cabeza N-proximal y una cola C-proximal. La proteína GyrA es del tipo "helix-loop-helix", compuesta por las hélices a3 y a4, que adopta diferentes conformaciones para permitir la apertura y el cierre de lugares determinados por los que debe pasar el DNA. Se cree que la girasa interactúa con el DNA a través de la hélice a4. Un detalle importante es que las zonas en que se producen las mutaciones frente a las quinolonas en la proteína GyrA (residuos 67, 81, 83 y 87) se sitúan cerca de la región de rotura-reunión de esta proteína y de la tirosina activa (Tyr-122), lo que confirma que las quinolonas interactúan con el complejo girasa-DNA (6). En E. coli y otras bacterias gramnegativas, como Neisseria gonorrhoeae y Pseudomonas aeruginosa, las quinolonas tienen como primera diana la DNA girasa (7-9). Sin embargo, estudios con Staphylococcus aureus y Streptococcus pneumoniae han rebatido la idea de que éste era un comportamiento general (10-15). En algunas bacterias grampositivas la situación es distinta: las primeras resistencias aparecen por mutaciones producidas en la topoisomerasa IV, mientras que las mutaciones en la girasa proporcionan resistencias adicionales (8, 9). De este modo, en S. aureus la topoisomerasa IV (cuyas subunidades se conocen como GrlA y GrlB en esta especie) es la primera diana de las quinolonas (10). En S. pneumoniae la actividad bactericida puede producirse sobre la girasa, la topoisomerasa IV o ambas, dependiendo de la estructura de la quinolona (14, 15). Este hecho señala una evidencia importante, ya que la relación entre estructura y actividad puede ser diferente para cada especie bacteriana. Debido a la aparición de resistencias, y buscando nuevas sustancias, las fluoroquinolonas se han estudiado profusamente durante los últimos años, fundamentalmente en S. pneumoniae. Se utiliza como el microorganismo de referencia puesto que se considera la principal causa de neumonía en la comunidad, además de estar implicado con frecuencia en exacerbaciones de bronquitis crónica y en meningitis, otitis media aguda y sinusitis (16). Así, según el orden de las consecutivas mutaciones QRDR en las topoisomerasas de mutantes resistentes seleccionados de S. pneumoniae, las quinolonas se pueden agrupar en tres clases (14, 15). El primer grupo, inicialmente identificado con ciprofloxacino, incluye también a levofloxacino, norfloxacino, pefloxacino y trovafloxacino, y se caracteriza por inducir mutaciones QRDR en la topoisomerasa IV antes que en la girasa, lo que sugiere que in vivo estos fármacos actúan preferentemente a través de la primera (12, 13, 17, 18). En cambio, un segundo grupo de quinolonas, compuesto por esparfloxacino, grepafloxacino, gatifloxacino, moxifloxacino y NSFQ-105 (homólogo del ciprofloxacino con un grupo 4-4-aminofenilsulfonil-1-piperacinilo en C7), produce mutaciones en la girasa antes que en la topoisomerasa IV, lo que señala como su diana principal a aquélla (15, 17, 19, 20). Finalmente, clinafloxacino y gemifloxacino actúan sobre ambas dianas, la girasa y la topoisomerasa IV (14, 21), pues aunque generan en primer lugar mutaciones QRDR en gyrA o gyrB, esta mutación simple se produce con poca frecuencia y en bajo grado, lo que indicaría que ambas contribuyen de forma importante en la acción del fármaco (14, 22). En conjunto estos resultados demuestran que la estructura de las quinolonas determina su modo de acción frente a S. pneumoniae (15, 19). Recientemente se han descrito en la bibliografía nuevas sustituciones en las posiciones C7 y C8 que cambian la primera diana de acción de las quinolonas de topoisomerasa IV a girasa o a ambas, y por tanto mejoran en gran medida la actividad antibacteriana en comparación con las quinolonas del grupo de ciprofloxacino (19, 23). En particular, el sustituyente de la posición 7 determina no sólo la potencia sino también la diana preferente de las fluoroquinolonas, como demuestra el trabajo publicado por Alovero y cols. (19). En él se estudia el mecanismo de acción de una nueva serie de fluoroquinolonas derivadas del ciprofloxacino por adición de un grupo bencenosulfonamida en el anillo piperacínico de la posición 7. Este trabajo establece que el aumento de la potencia que muestran estos compuestos, en especial el derivado NSFQ-105 con respecto a su homólogo, se asocia con la preferencia de la girasa como primera diana. A esta conclusión llegan tras observar que cepas mutantes en parC resistentes al ciprofloxacino permanecen sensibles al NSFQ-105, mientras que, por el contrario, cepas con mutaciones en gyrA son resistentes al NSFQ-105 y sensibles al ciprofloxacino. Esto demuestra que una modificación simple en el C7 de este compuesto es suficiente para hacer que la diana preferente de los derivados cambie de la topoisomerasa IV a la girasa. Dicho estudio confirma el modelo propuesto por Shen y cols. (24), que ya planteaba la idea de que el sustituyente en C7 está relacionado con las interacciones del fármaco y la enzima, conclusión que establecieron tras observar que las quinolonas con distintos sustituyentes en la citada posición mostraban in vitro diferentes actividades inhibidoras de la DNA girasa. También otros estudios muestran que la adición de un grupo metilo en el anillo piperacínico del C7 de las quinolonas aumenta en gran medida su actividad inhibidora en las topoisomerasas II de los mamíferos (25), lo que demuestra que el sustituyente de la posición 7 establece contactos críticos con la enzima que determina el reconocimiento de la diana bacteriana in vivo e in vitro. Por otro lado, el sustituyente de la posición 8 de las fluoroquinolonas parece favorecer la acción de éstas a través de la girasa en S. pneumoniae. A esta conclusión se llega al observar que esparfloxacino y clinafloxacino, que actúan sobre la girasa, tienen un átomo de flúor y un átomo de cloro como sustituyente en la posición C8, respectivamente, mientras que ciprofloxacino y trovafloxacino no presentan ningún sustituyente en dicha posición y ambas actúan a través de la topoisomerasa IV en esta especie. No se sabe si estas preferencias se deben a un aumento de la afinidad por la diana o a un aumento de la letalidad, como se ha descrito para una serie de derivados 8-metoxi del ciprofloxacino en mutantes de E. coli (23). Además, se cree que el sustituyente en C8 hace a las fluoroquinolonas especialmente activas frente a cepas resistentes por mutaciones en la girasa y la topoisomerasa IV en S. pneumoniae, como demuestra un estudio realizado con moxifloxacino, que tiene un grupo metoxi en la citada posición (20). Otro estudio reciente con gatifloxacino (que también posee un grupo metoxi en C8) y el citado microorganismo llega a la misma conclusión, estableciendo que el grupo 8-metoxi contribuye al aumento de la actividad antibacteriana tanto frente a cepas con mutaciones en las dianas como en cepas libres de mutaciones (26). Por último, cabe también comentar un trabajo muy reciente que demuestra que en S. aureus la presencia del grupo metoxi en C8 disminuye significativamente la tendencia al desarrollo de resistencias en comparación con las quinolonas con otros sustituyentes en esa posición (27). Conviene destacar que el hecho de que diferentes quinolonas tengan distintas dianas en la misma especie bacteriana tiene implicaciones importantes en la práctica clínica. Para varios autores (11, 12) lo más conveniente es que la quinolona actúe por igual en ambas dianas, puesto que las resistencias requieren la selección de dos mutaciones, una en cada diana, lo que ocurre con poca frecuencia. Este hecho explicaría la mejora en la actividad antineumocócica de clinafloxacino y gemifloxacino con respecto a sus precursoras. Así pues, sería muy interesante conocer con exactitud las estructuras moleculares que determinan la selección de la diana, pues ello daría paso a un diseño y una utilización dirigidos selectivamente a una u otra enzima, o a ambas a la vez. Por otra parte, el conocimiento del estado de las parC y gyrA QRDR en los agentes causantes de infecciones puede ser muy útil para evitar las resistencias (15). Por ejemplo, mutantes en parC y no en gyrA son resistentes al ciprofloxacino pero sensibles al esparfloxacino; en estos casos, esta última quinolona, desde el punto de vista exclusivamente microbiano, sería de elección. Por otro lado, las mutaciones QRDR en la girasa o la topoisomerasa IV no son los únicos mecanismos que desarrollan los microorganismos frente a la acción de los agentes antibacterianos, pues las resistencias a las quinolonas pueden estar condicionadas también por los procesos de transporte a través de la membrana (28). Así, el papel fundamental que ejercen los canales de las porinas en la difusión de las quinolonas hidrofílicas a través de la membrana externa de las bacterias gramnegativas quedó respaldado por la observación de que numerosos mutantes Mar (del inglés "multiple antibiotic resistant") que mostraban resistencias a estos agentes tenían en común la reducción del número de OmpF, la principal y mayor proteína de las porinas de la membrana externa de E. coli (29). Ello sugiere que las quinolonas hidrofílicas deben entrar en la célula bacteriana, al menos en parte, a través del canal de la porina OmpF. Estas cepas mutantes Mar de E. coli expresan resistencias codificadas por cromosomas frente a una gran variedad de antibióticos hidrofóbicos e hidrofílicos estructuralmente no relacionados. Entre los cambios que presentan estos mutantes Mar en la membrana externa destaca la importante reducción de la porina OmpF. Estas observaciones implican que la reducción de OmpF requiere mutaciones en la región génica marA (30), es decir, que mutaciones en el locus marA confieren resistencia a varios grupos de antibióticos, entre ellos las quinolonas, por un mecanismo que reduce la permeabilidad al disminuir la expresión de la OmpF. Las mutaciones que afectan a la permeabilidad confieren bajos grados de resistencia a las quinolonas (aumento de 2 a 4 diluciones log2 de la CMI) y producen habitualmente resistencias cruzadas con otros antibióticos estructuralmente no relacionados (29). Como norma general se considera que un tamaño voluminoso, la carga negativa y un aumento de la hidrofobicidad retrasan la penetración de los antibacterianos en los microorganismos gramnegativos a través de los canales de las porinas (31). Por otro lado, las moléculas hidrofóbicas parecen utilizar una ruta alternativa de difusión a través de lipopolisacáridos (32, 33). Algunos autores (34) han demostrado que la hidrofobicidad de los compuestos parece tener relevancia tanto en grampositivos como en gramnegativos, mientras que el elevado peso molecular es un factor limitante sólo para los microorganismos gramnegativos. Posteriormente se conoció la implicación en la resistencia a las quinolonas del tercer mecanismo señalado: la bomba de expulsión activa. Las quinolonas son sustratos de este transportador que actúa extrayendo el fármaco desde el medio intracelular al extracelular e impide así la acumulación. El sistema de expulsión se sitúa en la membrana interna de los microorganismos y cataliza un proceso dependiente de energía ligado a un gradiente de protones (35). En E. coli este sistema de transporte, denominado AcrAB, es una bomba de flujo multifármaco que extrae de la célula una amplia variedad de antibióticos, incluidas las quinolonas y otras sustancias (36). Este sistema AcrAB está compuesto por el transportador AcrB y la proteína periplásmica accesoria AcrA. Se cree que AcrA, de forma alargada, aproxima las membranas externa e interna, formando un trímero que interacciona con el monómero AcrB, bombeando así una amplia variedad de sustancias, presumiblemente a través del canal TolC de la membrana externa (37). El sistema AcrAB está codificado por los genes acrAB y parece tener una función fisiológica en E. coli, que consiste en proteger a la célula frente a sales biliares y ácidos grasos, tóxicos habituales de su entorno fisiológico (38). Es importante destacar que la expresión de los genes acrAB aumenta de forma considerable en los mutantes Mar, lo que implica que el locus MarA de E. coli regula no sólo la expresión de la porina OmpF, sino también la expresión de la bomba AcrAB (38). Es decir, los mutantes Mar presentan resistencias debido a una disminución de la permeabilidad de la membrana externa y a una importante expulsión activa a través de la membrana interna, lo que repercute en una disminución de la entrada del fármaco y un aumento de su salida, y como consecuencia una disminución de la sensibilidad a éste. Un esquema de este proceso de entrada y salida del antibiótico se representa en la Fig. 1.
Figura 1. Entrada y expulsión de las quinolonas en E. coli. Q: quinolona. PmF: fuerza protón motriz. Entre las bacterias grampositivas se han identificado bombas de flujo en Bacillus subtilis (Bmr) (39) y S. aureus (NorA) (40, 41). El gen bmr codifica el transportador multifármaco Bmr de B. subtilis. Este transportador, al igual que el AcrAB de E. coli, bombea al exterior de la célula compuestos tóxicos de estructura diversa (42). Se ha comprobado que la sobrexpresión del gen bmr en este microorganismo genera resistencias elevadas a diversos fármacos, incluidas las fluoroquinolonas. Este transporte puede revertirse con reserpina, que se comporta como un inhibidor del transportador Bmr (43). En cuanto a S. aureus, algunas de las resistencias pueden asociarse a un aumento de la transcripción del gen norA (41). El gen norA codifica las proteínas de membrana participantes en los procesos de expulsión, que pueden dar lugar a resistencias en S. aureus y también en E. coli (41, 44). Destaca, pues, que el grado de expresión de este gen determina la sensibilidad tanto de bacterias grampositivas como gramnegativas. El efecto de la expresión del gen norA sobre la actividad de las quinolonas depende de su afinidad por el transportador NorA, que se manifiesta fundamentalmente para las fluoroquinolonas hidrofílicas y confiere bajo grado de resistencia (41). Cabría destacar que la proteína Bmr de B. subtilis es muy similar a la proteína NorA de S. aureus, tanto en estructura como funcionalmente (39). En fecha reciente también se ha descrito un mecanismo de expulsión como causa de bajos grados de resistencia a las quinolonas en S. pneumoniae (45). A la bomba causante de esta acción se la ha denominado bomba PmrA (del inglés "pneumococcal multidrug resistance protein") (46). Se trata también de una proteína asociada a la membrana que se inhibe con reserpina, aunque las bases genéticas de este mecanismo no se conocen. Se han descrito otros transportadores similares que afectan a la sensibilidad a las quinolonas en otras especies bacterianas, como por ejemplo el gen mexABoprK de P. aeruginosa que proporciona resistencia al ciprofloxacino y al ácido nalidíxico (47). En resumen, la expresión de los transportadores de expulsión puede determinar la sensibilidad antimicrobiana y está reconocida como causa de resistencia de bajo grado a las quinolonas. Además, algunas quinolonas presentan una baja afinidad por estos transportadores, lo que contribuye a aumentar su potencia (9). Por otro lado, el hecho de que este efecto es dependiente de la concentración determina, al menos en parte, que las fluoroquinolonas presenten mayor letalidad a concentraciones bajas, y ello dificulta que la bacteria desarrolle mutaciones en los cromosomas que puedan conducir a altos grados de resistencia (48). Dada la trascendencia de estos hallazgos se ha intentado establecer una relación entre los procesos de expulsión y la estructura de las quinolonas. Varios autores (41, 44) consideran que la hidrofilia es el principal determinante de los altos grados de resistencia, especialmente en microorganismos con proteínas NorA. Estudios en S. aureus (49) han demostrado que la hidrofilia de las quinolonas no es un factor exclusivo, y han argumentado que la actividad sobre cepas resistentes se correlaciona mejor con el volumen del sustituyente en C7 y el volumen y la hidrofobia del sustituyente en C8. Brenwald y cols. (45) reconsideraron la idea de la hidrofilia y demostraron que las fluoroquinolonas hidrofóbicas son sustratos pobres de la bomba de expulsión neumocócica. Concluyendo se puede afirmar que los mecanismos de resistencia a las fluoroquinolonas consisten en mutaciones en los genes que codifican la DNA girasa y la topoisomerasa IV, alteraciones en la permeabilidad de la membrana y flujo activo de los antimicrobianos desde las células al exterior. Este proceso de expulsión es importante ya que puede permitir que las bacterias sobrevivan durante un corto periodo de tiempo y desarrollen resistencias vía mutaciones en los lugares clave de los genes de las dianas de las quinolonas. Los tres tipos de mecanismos de resistencia pueden manifestarse solos o en combinación, si bien se cree que el aumento en el grado de resistencia a las quinolonas in vivo se debe a varios mecanismos simultáneos o secuenciales. Correspondencia: Raquel Taléns-Visconti, Dpto. Farmacia y Tecnología Farmacéutica, Facultad de Farmacia, Universidad de Valencia, Avda. V.A. Estellés s/n, 46100 Burjassot (Valencia). BIBLIOGRAFÍA 1. Nakamura S. Mechanisms of quinolone resistance. J Infect Chemother 1997; 3: 128-138. 2. Wolfson, J.S., Hooper, D.C. Fluoroquinolone antimicrobial agents. Clin Microbiol Rev 1989; 2: 378-424. 3. Yoshida, H., Bogaki, M., Nakamura, M., Nakamura, S. Quinolone resistance-determining region in the DNA gyrase gyrA gene of Escherichia coli. Antimicrob Agents Chemother 1990; 34: 1271-1272. 4. Yoshida, H., Bogaki, M., Nakamura, M., Yamanaka, L.M., Nakamura, S. Quinolone resistance-determining region in the DNA gyrase gyrB gene of Escherichia coli. Antimicrob Agents Chemother 1991; 35: 1647-1650. 5. Heisig P. Genetic evidence for a role of parC mutations in development of high-level fluoroquinolone resistance in Escherichia coli. Antimicrob Agents Chemother 1996; 40: 879-855. 6. Morais Cabral, J.H., Jackson, A.P., Smith, C.V., Shikotra, N., Maxwell, A., Liddington, R.C. Crystal structure of the breakage-reunion domain of DNA gyrase. Nature 1997; 388: 903-906. 7. Akasaka, T., Onodera, Y., Tanaka, M., Sato, K. Cloning, expression, and enzymatic characterization of Pseudomonas aeruginosa topoisomerase IV. Antimicrob Agents Chemother 1999; 43: 530-536. 8. Drlica, K., Zhao, X. DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol Mol Biol Rev 1997; 61: 377-392. 9. Hooper, D.C. Quinolone mode of action. Drugs 1995; 49 (Suppl. 2): 10-15. 10. Ferrero, L., Cameron, B., Manse, B. y cols. Cloning and primary structure of Staphylococcus aureus DNA topoisomerase IV: A primary target of fluoroquinolones. Mol Microbiol 1994; 13: 641-653. 11. Ng, E.Y.W., Trucksis, M., Hooper, D.C. Quinolone resistance mutations in topoisomerase IV: Relationship to the flqA locus and genetic evidence that topisomerase IV is the primary target and DNA gyrase is the secondary target of fluoroquinolones in Staphylococcus aureus. Antimicrob Agents Chemother 1996; 40: 1881-1888. 12 Pan, X.-S., Ambler, J., Mehtar, S., Fisher, L.M. Involvement of topoisomerase IV and DNA gyrase as ciprofloxacin targets in Streptococcus pneumoniae. Antimicrob Agents Chemother 1996; 40: 2321-2326. 13. Pan, X.-S., Fisher, L.M. Cloning and characterization of the parC and parE genes of Streptococcus pneumoniae encoding DNA topoisomerase IV: Role in fluoroquinolone resistance. J Bacteriol 1996; 178: 4060-4069. 14. Pan, X.-S., Fisher, L.M. DNA gyrase and topoisomerase IV are dual targets of clinafloxacin action in Streptococcus pneumoniae. Antimicrob Agents Chemother 1998; 42: 2810-2816. 15. Pan, X.-S., Fisher, L.M. Targeting of DNA gyrase in Streptococcus pneumoniae by sparfloxacin: Selective targeting of gyrase or topoisomerase IV by quinolones. Antimicrob Agents Chemother 1997; 41: 471-474. 16. Barlett, J.G., Grundy, L.M. Community-acquired pneumonia. N Engl J Med 1995; 333: 1618-1624. 17. Fukuda, H., Hiramatsu, K. Primary targets of fluoroquinolones in Streptococcus pneumoniae. Antimicrob Agents Chemother 1999; 43: 410-412. 18. Muñoz, R., De la Campa, A.G. ParC subunit of DNA topoisomerase IV of Streptococcus pneumoniae is a primary target of quinolones and cooperates with DNA gyrase A subunit in forming resistance phenotype. Antimicrob Agents Chemother 1996; 40: 2252-2257. 19. Alovero, F.L., Pan, X.-S., Morris, J.E., Manzo, R.H., Fisher, L.M. Engineering the specificity of antibacterial fluoroquinolones: Benzenesulfonamide modifications at C-7 of ciprofloxacin change its primary target in Streptococcus pneumoniae from topoisomerase IV to gyrase. Antimicrob Agents Chemother 2000; 44: 320-325. 20. Pestova, E., Millichap, J.J., Noskin, G.A., Peterson, L.R. Intracellular targets of moxifloxacin: A comparison with other fluoroquinolones. J. Antimicrob Chemother. 2000; 45: 583-590. 21. Heaton, V.J., Goldsmith, C.G., Ambler, J.E., Fisher, L.M. Activity of gemifloxacin against penicillin- and ciprofloxacin-resistant Streptococcus pneumoniae displaying topoisomerase- and efflux-mediated resistance mechanisms. Antimicrob Agents Chemother 1999; 43: 2998-3000. 22. Heaton, V.J., Ambler, J.E., Fisher, L.M. Potent antineumococcal activity of gemifloxacin is associated with dual targeting of gyrase and topoisomerase IV, an in vivo target preference for gyrase, and enhanced stabilization of cleavage complexes in vitro. Antimicrob Agents Chemother 2000; 44: 3112-3117. 23. Zhao, X., Xu, C., Domagala, J., Drlica, K. DNA topoisomerase targets of the fluoroquinolones: A strategy for avoiding bacterial resistance. Proc Natl Acad Sci USA 1997; 94: 13991-13996. 24. Shen, L.L., Mitscher, L.A., Sharma, P.N. y cols. Mechanism of inhibition of DNA gyrase by quinolone antibacterials: A cooperative drug DNA binding model. Biochemistry 1989; 28: 3886-3894. 25. Gootz, T.D., McGuirk, P.R., Moynihan, M.S., Haskell, S.L. Placement of alkyl substituents on the C-7 piperazine ring of fluoroquinolones: Dramatic effects on mammalian topoisomerase II and DNA gyrase. Antimicrob Agents Chemother 1994; 38: 130-133. 26. Fukuda, H., Kishii, R., Takei, M., Hosaka, M. Contributions of the 8-methoxy group of gatifloxacin to resistance selectivity, target preference, and antibacterial activity against Streptococcus pneumoniae. Antimicrob Agents Chemother 2001; 45: 1649-1653. 27. Dalhoff, A. Comparative in vitro and in vivo activity of the C-8 methoxy quinolone moxifloxacin and the C-8 chlorine quinolone BAY y 3118. Clin Infect Dis. 2001; 15; 32 (Suppl. 1): S16-22. 28. Nikaido, H. Preventing drug access to targets: Cell surface permeability barriers and active efflux in bacteria. Semin Cell Dev Biol 2001; 12: 215-23. 29. Cohen, S.P., McMurry, L.M., Hooper, D.C., Wolfson, J.S., Levy, S.B. Cross-resistance to fluoroquinolones in multiple-antibiotic-resistant (Mar) Escherichia coli selected by tetracycline or chloramphenicol: Decreased drug accumulation associated with membrane changes in addition of OmpF reduction. Antimicrob Agents Chemother 1989; 33: 1318-1325. 30. Cohen, SP., McMurry, L.M., Levy, S.B. marA locus causes decreased expression of OmpF porin in multiple-antibiotic resistant (Mar) mutants of Escherichia coli. J Bacteriol 1988; 170: 5416-5422. 31. Hancock, R.E.W. Role of porin in outer membrane permeability. J Bacteriol 1987; 169: 929-934. 32. Chapman, J.S., Georgopapadakou, N.H. Routes of quinolone permeation in Escherichia coli. Antimicrob Agents Chemother 1988; 32: 438-442. 33. Nikaido, H., Thanassi, D.G. Penetration of lipophilic agents with multiple protonation sites into bacterial cells: Tetracyclines and fluoroquinolones as examples. Antimicrob Agents Chemoter 1993; 37: 1393-1399. 34. Bazile, S., Moreau, N., Bouzard, D., Essiz, M. Relationships among antibacterial activity, inhibition of DNA gyrase, and intracellular accumulation of 11 fluoroquinolones. Antimicrob Agents Chemother 1992; 36: 2622-2627. 35. Cohen, S.P., Hooper, D.C., Wolfson, J.S., Souza, K.S., McMurry, L.M., Levy, S.B. Endogenous active efflux of norfloxacin in susceptible Escherichia coli. Antimicrob Agents Chemother 1988; 32: 1187-1191. 36. Nikaido, H. Multidrug efflux pumps of gram-negative bacteria. J Bacteriol 1996; 178: 5853-5859. 37. Nikaido, H., Zgurskaya, H.I. AcrAB and related multidrug efflux pumps of Escherichia coli. J Mol Microbiol Biotechnol 2001; 3: 215-218. 38. Ma, D., Cook, D.N., Alberti, M., Pon, N.G., Kikaido, H., Hearst, J.E. Genes acrA and acrB encode a stress-induced efflux system of Escherichia coli. J Mol Microbiol 1995; 16: 45-55. 39. Neyfakh, A.A. The multidrug efflux transporter of Bacillus subtilis is a structural and functional homolog of the Staphylococcus NorA protein. Antimicrob Agents Chemother 1992; 36: 484-485. 40. Ubukata, K., Itoh-Yamashita, N., Konno, M. Cloning and expression of the norA gene for fluoroquinolone resistance in Staphylococcus aureus. Antimicrob Agents Chemother 1989; 33: 1535-1539. 41. Yoshida, H., Bogaki, M., Nakamura, S., Ubukata, K., Konno, M. Nucleotide sequence and characterization of the Staphylococcus aureus norA gene, which confer resistance to quinolones. J Bacteriol 1990; 172: 6942-6949. 42. Neyfakh, A.A., Bidnenko, V.E., Chen, L.B. Efflux-mediated multidrug resistance in Bacillus subtilis: Similarities and dissimilarities with the mammalian system. Proc Natl Acad Sci USA 1991; 88: 4781-4785. 43. Ahmed, M., Borsch, C.M., Neyfakh, A.A., Schuldiner, S. Mutants of the Bacillus subtilis multidrug transporter Bmr with altered sensitivity to the antihypertensive alkaloid reserpine. J Biol Chem 1993; 268: 11086-11089. 44. Ng, E.Y.W., Trucksis, M., Hooper, D.C. Quinolone resistance mediated by norA: Physiologic characterization and relationship to flqB, a quinolone resistance locus on the Staphylococcus aureus chromosome. Antimicrob Agents Chemother 1994; 38: 1345-1355. 45. Brenwald, N.P., Gill, M.J., Wise, R. Prevalence of a putative efflux mechanism among fluoroquinolone-resistant clinical isolates of Streptococcus pneumoniae. Antimicrob Agents Chemother 1998; 42: 2032- 2035. 46. Gill, M.J., Brenwald, N.P., Wise, R. Identification of an efflux pump gene, pmrA, associated with fluoroquinolone resistance in Streptococcus pneumoniae. Antimicrob Agents Chemother 1999; 43: 187-189. 47. Poole, K., Krebes, K., McNally, C., Neshat, S. Multiple antibiotic resistance in Pseudomonas aeruginosa: Evidence for involvement of an efflux operon. J. Bacteriol. 1993; 175: 7363-7372. 48. Pestova, E., Beyer, R., Cianciotto, N.P., Noskin, G.A., Peterson, L.R. Contribution of topoisomerase IV and DNA gyrase mutations in Streptococcus pneumoniae to resistance to novel fluoroquinolones. Antimicrob Agents Chemother 1999; 43: 2000-2004. 49. Takenouchi, T., Tabata, F., Iwata, Y., Hanzawa, H., Sugawara, M., Ohya, S. Hydrophilicity of quinolones is not an exclusive factor for decrease activity in efflux-mediated resistant mutants of Staphylococcus aureus. Antimicrob Agents Chemother 1996; 40: 1835-1842.
|