- Service, R.F. Antibiotic that resist resistance. Science 1995; 270: 724-727.
- Appelbaum, P.C. Antimicrobial resistance in Streptococcus pneumoniae: An overview. Clin Infect Dis 1992; 15: 77-83.
- Doern, G.V., Brueggemann, A., Holley, H.P. Jr., Rauch, A.M. Antimicrobial resistance of Streptococcus pneumoniae recovered from outpatients in the United Sates during the winter months of 1994 to 1995: Results of a 30-center national surveillance study. Antimicrob Agents Chemother 1996; 40: 1208-1213.
- Tomasz, A. Antibiotic resistance in Streptococcus pneumoniae. Clin Infect Dis 1997; 24: S85-S88.
- McDougal, L.K., Rasheed, J.K., Biddle, J.W., Tenover, F.C. Identification of multiple clones of extended-spectrum cephalosporin-resistant Streptococcus pneumoniae isolates in the United States. Antimicrob Agents Chemother 1995; 39: 2282-2288.
- Gould, H.S., Moellering, R.C. Antimicrobial-drug resistance. N Engl J Med 1996; 335: 1445-1453.
- Neu, H. The crisis in antibiotic resistance. Science 1992; 257: 1064-1073.
- Panlilio, A.L., Culver, D.H,. Gaynes, R.P., Banerjee, S., Henderson, T.S., Tolson, J.S., Martone, W.J. Methilicin-resistant Staphylococcus aureus in U.S. hospitals, 1975-1991. Infect Control Hosp Epidemiol 1992; 13: 582-586.
- Wood, A.J.J. Antimicrobial drug resistance. Drug Therapy 1996; 335: 1445-1453.
- Hiramatsu, K., Hanaki, H., Ino, T., Yabuta, K., Oguri, T., Tenover, F.C. Methicilin-resistant Staphylococcus aureus clinical strain with reduced vancomycin susceptibility. J Antimicrob Chemother 1997; 40: 135-136.
- Wallace, R.J. Jr., Steingrube, V.A., Nash, D.R., Hollis, D.G., Flanagan, C., Brown, B.A., Labidi, A., Weaver, R.E. BRO beta-lactamases of Branhamella catarrhalis and Moraxella subgenus Moraxella, including evidence for chromosomal beta-lactamase transfer by conjugation in B. catarrhalis, M. nonliquefaciens and M. lacunata. Antimicrob Agents Chemother 1989; 33: 1845-1854.
- Tenover, F. Novel and emerging mechanisms of antimicrobial resistance in nosocomial pathogens. Am J Med 1991; 91 (3B): 76S-81S.
- Glynn, M.K., Bopp, C., Dewit, W., Dabney, P., Mokhtar, M., Angulo, F. Emergence of multidrug-resistant Salmonella enterica serotype typhymurium DT104 infections in the United States. N Engl J Med 1998; 338: 1333-1338.
- Neu, H.C. Contribution of beta-lactamases to bacterial resistance and mechanisms to inhibit beta-lactamases. Am J Med 1985; 79 (5B): 2-12.
- Rao, G.G. Risk factors for the spread of antibiotic-resistant bacteria. Drugs 1998; 55: 323-330.
- Houghten, R.A., Pinilla, C., Blondelle, S.E., Appel, J.R., Dooley, C.T., Cuervo, J.H. Generation and use of synthetic peptide combinatorial libraries for basic research and drug discovery. Nature 1991; 354: 84-86.
- Wilson and Czarnik (Eds.). Combinatorial chemistry. Synthesis and application. John Willey and Sons, New York 1997.
- Pérez-Payá, E. Peptidotecas combinatoriales sintéticas. En: Andreu, D., Rivas, L. (Eds.). Péptidos en Biología y Medicina. Col. Nuevas Tendencias, CSIC, Madrid 1997; 238-254.
- Blondelle, S.E., Pérez-Payá, E., Houghten, R.A. Synthetic combinatorial libraries: Novel discovery strategy for identification of antimicrobial agents. Antimicrob Agents Chemother 1996; 40: 1067-1071.
- Blondelle, S.E., Houghten, R.A. Novel antimicrobial compounds identified using synthetic combinatorial library technology. TIBTECH 1996; 14: 60-65.
- Kay, Winter and McCafferty (Eds.). Phage display of peptides and proteins. A laboratory manual. Academic Press, New York 1996.
- Geysen, H.M., Rodda, S.J., Mason, T.L. A priori delineation of a peptide which mimics a discontinuous antigenic determinant. Mol Immunol 1986; 23: 709-715.
- Lam, K.S., Salmon, S.E., Hersh, E.M., Hruby, V.J., Kazmierski, W.M., Knapp, R.J. A new type of synthetic peptide library for identifying ligand-binding activity. Nature 1991; 354: 82-84.
- Houghten, R.A., Appel, J.R., Blondelle, S.E., Cuervo, J.H., Dooley, C.T., Pinilla, C. The use of synthetic peptide combinatorial libraries for the identification of bioactive peptides. Biotechniques 1992; 13: 412-421.
- Houghten, R.A. General method for the rapid solid-phase synthesis of large numbers of peptides: Specificity of antigen-antibody interaction at the level of individual amino acids. Proc Natl Acad Sci USA 1985; 82: 5131-5135.
- Pinilla, C., Appel, J.R., Blanc, P., Houghten, R.A. Rapid identification of high affinity peptide ligands using positional scanning synthetic peptide combinatorial libraries. Biotechniques 1992; 13: 901-905.
- Zasloff, M. Antibiotic peptides as mediators of innate inmunity. Curr Opin Immunol 1992; 4 (1): 3-7.
- Saberwal, G., Nagarj, R. Cell-lytic and antibacterial peptides that act by perturbing the barrier function of membranes: Facets of their conformational features, structure-function correlations and membrane-perturbing abilities. Biochem Biophys Acta 1994; 1197: 109-131.
- Jawetz, E. Polymixin, colistin and bacitracin. Pediatr Clin N Am 1961; 8: 1057-1071.
- Verhoef, J.C., Bodde, H.E., Deboer, A.G., Bouwstra, J.A., Junginger, F.W., Merkus, H.M., Breimer, D.D. Transport of peptide and protein drugs across biological membranes. Eur J Drug Metab Pharmacokinet 1990; 15: 83-93.
- Blondelle, S.E., Takahashi, E., Dinh, K.T., Houghten, R.A. The antimicrobial activity of hexapeptides derived from synthetic combinatorial libraries. J Appl Bacteriol 1995; 78: 39-46.
- Blondelle, S.E., Takahashi, E., Dinh, K.T., Houghten, R.A., Pérez-Payá, E. Rapid identification of compounds with enhanced antimicrobial activity by using conformationality defined combinatorial libraries. Biochem J 1996; 313: 141-147.
- Hong, S.Y., Oh, J.E., Kwon, M.Y., Choi, M.J., Lee, J.H., Lee, B.L., Moon, H.M., Lee, K.H. Identification and characterization of novel antimicrobial decapeptides generated by combinatorial chemistry. Antimicrob Agents Chemother 1998; 42: 2534-2541.
- Blondelle, S.E., Takahashi, E., Weber, P.A., Houghten, R.A. Identification of antimicrobial peptides identified using combinatorial libraries made up of unnatural amino acids. Antimicrob Agents Chemother 1994; 38: 2280-2286.
- Bunin, B.A., Ellman, M.H. A general and expedient method for the solid synthesis of 1,4-benzodiazepine derivatives. J Am Chem Soc 1992; 114: 10997-10998.
- Simon, R.J., Kania, R.S., Zuckermann, R.N., Huebner, V.D., Jewell, D.A., Banville, S., Ng, S., Wang, L., Rosenberg, S., Marlowe, C.K., Spellmeyer, D.C., Tan, R., Frankel, A.D., Santi, D.V., Cohen, F.E., Bartlett, P.A. Peptoids: A molecular approach to drug discovery. Proc Natl Acad Sci USA 1992; 89: 9367-9371.
- Cho, Y.C., Moran, E.J., Cherry, S.R., Stephans, J.C., Fodor, S.P.A., Adams, C.L., Sundaram, A., Jacobs, J.W., Schultz, P.G. An unnatural biopolymer. Science 1993; 261: 1303-1305.
- DeWitt, S.H., Kiely, J.S., Stankovic, C.J., Schroeder, M.C., Cody, D.M.R., Pavia, M.R. "Diversomers": An approach to nonpeptide, nonoligomeric chemical diversity. Proc Natl Acad Sci USA 1993; 90: 6909-6913.
- Chen, C.L., Ahlberg-Randall, L.A., Miller, R.B., Jones, A.D., Kurth, L. "Analogous" organic synthesis of small-compound libraries: Validation of combinatorial chemistry in small molecule synthesis. J Am Chem Soc 1994; 116: 2661-2662.
- Carell, T., Wintner, E.A., Bashir-Hashemi, A., Rebek, J. Jr. A novel procedure for the synthesis of libraries containing small organic molecules. Angew Chem Int Ed Engl 1994; 33: 2059-2061.
- Silen, J.L., Lu, A.T., Solas, D.W., Gore, M.A., McLean, D., Shah, N.H., Coffin, J.M., Bhinderwala, N.S., Wang, Y., Tsutsui, K.T., Look, G.C., Campbell, D.A., Hale, R.L., Navre, M., DeLuca-Flaherty, C.R. Screening for novel antimicrobials from encoded combinatorial libraries by using a two-dimensional agar format. Antimicrob Agents Chemother 1998; 42: 1447-1453.
Química combinatoria en biomedicina: una nueva estrategia en el desarrollo de antibióticos
M. Vilar, T. Carbonell y E. Pérez-Payá Departamento de Bioquímica y Biología Molecular, Universidad de Valencia, c/ Dr. Moliner no 50, 46100 Burjassot (Valencia).Tras varias décadas de uso abusivo e indiscriminado de los antibióticos se ha llegado a una situación en la que se han seleccionado cepas bacterianas resistentes y que, por tanto, tienen incrementada la virulencia del patógeno en su mecanismo de infección. La propagación de estas cepas resistentes a los antibióticos hoy conocidos (unos 160 compuestos) (1) es un grave problema y se apunta como una de las principales causas de muerte en el próximo milenio. Basta con mencionar que enfermedades como la tuberculosis, la fiebre tifoidea, la meningitis, la neumonía y las septicemias son cada vez más frecuentes en zonas donde se pensaba que ya estaban erradicadas; las mismas bacterias que durante mucho tiempo habían sido controladas con antibióticos, son ahora invulnerables. Es por esta situación de alarma por lo que surge la necesidad de desarrollar urgentemente nuevos compuestos activos. Un ejemplo de esta situación lo tenemos en Streptococcus pneumoniae, que en los años 1940 era sensible a la penicilina; concentraciones tan bajas como 0,01 �g/ml no sólo inhibían su crecimiento sino que, además, provocaban su lisis. En cambio, tras varios años de tratamiento con penicilina, la concentración mínima inhibitoria (CMI) pasaba a ser mayor de 4-8 �g/ml (ya en la década de 1970). Estas cepas de S. pneumoniae no sensibles a la penicilina se han distribuido por todo el mundo y, más aún, la mayoría de ellas han desarrollado también resistencia a otros antibióticos como la eritromicina, la tetraciclina, la asociación trimetoprima-sulfametoxazol y el cloranfenicol (2-6). Por todo esto, actualmente es uno de los patógenos más peligrosos y temidos.
También se han descrito cepas de enterococos resistentes a las penicilinas, los aminoglucósidos y las ampicilinas, y más recientemente a la vancomicina (7). Por otra parte, alrededor del 90% de las cepas clínicas de Staphylococcus aureus son capaces de sintetizar una betalactamasa (penicilinasa) que les confiere resistencia a varios betalactámicos (particularmente a las penicilinas) (8, 9).
En el caso de S. aureus, para controlar su infección se utilizaban sulfonamidas (en los años 1930) y bencilpenicilina (en los años 1940), pero el problema de la resistencia comenzó a ponerse de manifiesto a finales de la década de 1950 porque el uso continuado de bencilpenicilina había actuado como factor de selección de cepas resistentes que sintetizaban penicilinasa. Y fue en 1959 cuando se introdujo la meticilina, una penicilina semisintética que no es inactivada por la penicilinasa; sin embargo, tan pronto como en 1961 ya se aisló en Europa la primera cepa de S. aureus resistente a este antibiótico (SARM, S. aureus resistente a meticilina). Con el uso extensivo de éste y otros antimicrobianos durante muchos años, diferentes cepas de SARM han desarrollado resistencia a casi todas las clases de antibióticos, excepto a los glucopéptidos, que parecían ser los únicos compuestos capaces de actuar frente a todas las cepas de SARM. Pero la voz de alarma se disparó cuando se describió la primera de estas cepas resistente a la vancomicina (glucopéptido), aunque el mecanismo de resistencia todavía no está bien descrito (10) puesto que las bacterias cuentan con multitud de estrategias que les permiten desarrollar resistencia, como por ejemplo mutaciones de los genes que codifican para las proteínas diana, conjugación con cepas resistentes, genes que confieren resistencia codificados por transposones, intercambio de material genético exógeno, etc.
Con todo ello, si la resistencia a la vancomicina se propagase no sería descabellado pensar en una vuelta a la era preantibiótica, cuando las infecciones por S. aureus y S. pneumoniae eran mortales. Aunque las infecciones por SARM se producen mayoritariamente en ambientes hospitalarios, no están restringidas únicamente a éstos sino que parecen extenderse a otros entornos.
Finalmente, y por mencionar otras bacterias significativas que también han desarrollado resistencia a los antibióticos, se han descrito cepas de Haemophilus influenzae resistentes a la ampicilina, el cloranfenicol y la asociación trimetoprima-sulfametoxazol (7), de Neisseria meningitidis resistentes a la penicilina (7), de Moraxella catarrhalis resistentes a la ampicilina y la amoxicilina (11), y de Salmonella typhy resistentes al cloranfenicol, la ampicilina y trimetoprima-sulfametoxazol (12, 13). Además, también habría que mencionar las bacterias gramnegativas multirresistentes, como son los géneros Klebsiella, Serratia, Proteus, Enterobacter y Escherichia (12), y Pseudomonas aeruginosa (14).
Por desgracia, el desarrollo de nuevos antibióticos está siendo un proceso mucho más lento que el de la propagación de estas cepas, que se ve favorecido, a su vez, por la masificación, la higiene deficiente y los escasos controles de infección en muchos hospitales (15). Con todo ello se hace patente que el problema de la resistencia a los antibióticos es una realidad y que está naciendo una nueva "era de los antibióticos"; las infecciones bacterianas, uno de los campos donde la química farmacológica más éxitos ha obtenido, y que parecía ya agotado, está en pleno renacimiento. En paralelo con esta revitalización han aparecido nuevas técnicas de síntesis, ensayo e identificación de compuestos con posible actividad antibiótica, a partir de millones de moléculas diferentes que, sin duda, agilizará y facilitará el proceso de identificación de nuevos antibióticos y posiblemente sea la única vía para encontrar las "penicilinas" del siglo xxi. Esta nueva técnica es la química combinatoria y posibilita la síntesis de quimiotecas, tanto de péptidos como de moléculas orgánicas no peptídicas, consiguiendo una diversidad química elevada y la realización del ensayo biológico en un tiempo récord.
QUIMIOTECAS COMBINATORIAS
Una quimioteca se define como una colección de compuestos, desde decenas hasta millones, que se preparan simultáneamente por medio de síntesis múltiple. Estas quimiotecas destacan por contener todas las combinaciones posibles de una misma clase de compuestos. En 1991, la introducción de las quimiotecas permitió un importante avance práctico en la investigación farmacéutica (16). Estas quimiotecas se han utilizado para la identificación de agonistas y antagonistas de receptores opiáceos y de inhibidores de enzimas, para estudios de unión entre ligando y proteína y de interacción de antígeno y anticuerpo, y más recientemente para la identificación de antimicrobianos (17-20). En la actualidad se han puesto a punto diferentes estrategias sintéticas para preparar conjuntos de moléculas de alta diversidad química en condiciones que permitan su utilización en ensayos biológicos. Todas las quimiotecas elaboradas hasta ahora se basan en la síntesis de péptidos en fase sólida y se pueden dividir en tres categorías. La primera utiliza las técnicas de biología molecular, donde secuencias de péptidos son "presentadas" en la superficie de las proteínas de cubierta de fagos filamentosos, técnica denominada phage display (17, 18, 21). El principal inconveniente es que como se basa en un sistema biológico no se pueden incorporar a las secuencias D-aminoácidos ni aminoácidos no naturales, que aumentarían la diversidad y la versatilidad de las técnicas combinatorias. La segunda categoría de quimiotecas implica el uso de síntesis química y ensayo biológico posterior sobre soportes sólidos: la síntesis de los compuestos se realiza acoplada a un soporte sólido y, al finalizar la síntesis, las moléculas quedan unidas a éste; así, los ensayos se realizan con las moléculas inmovilizadas en el soporte (22, 23), lo que limita el tipo y la calidad del ensayo. La tercera categoría, que opinamos es la más interesante para su aplicación en el campo antimicrobiano, se basa en quimiotecas sintéticas solubles, donde los compuestos sintetizados sobre el soporte sólido son posteriormente escindidos por métodos químicos (16, 24). Una vez en disolución se pueden aplicar a cualquier tipo de ensayo biológico que se diseñe convenientemente y la información obtenida es mucho más representativa. Además, por ser esta categoría la más utilizada en la obtención de quimiotecas de compuestos orgánicos y con la que mejores resultados se han obtenido en la identificación de antibióticos, describiremos con cierto detalle el procedimiento sintético y sus posibilidades de aplicación.
Quimiotecas sintéticas solubles
Las quimiotecas sintéticas solubles se preparan mediante síntesis múltiple en fase sólida (25). La primera unidad que se incorpora quedaría covalentemente unida a un soporte sólido, que es un polímero en forma de perla (llamado resina) que contiene grupos reactivos donde puede crecer el péptido. Posteriormente las moléculas son escindidas del soporte y el péptido (o molécula orgánica) así liberado queda en disolución. Existen dos tipos de descodificación para la identificación de compuestos activos: a) un proceso iterativo que incluye sucesivos pasos de síntesis, rastreo, selección de los compuestos más activos y con los seleccionados repetición de los mismos pasos hasta encontrar la secuencia más activa (Fig. 1), y b) un proceso de rastreo posicional, donde se pueden encontrar las secuencias activas en un solo ensayo (Fig. 2).
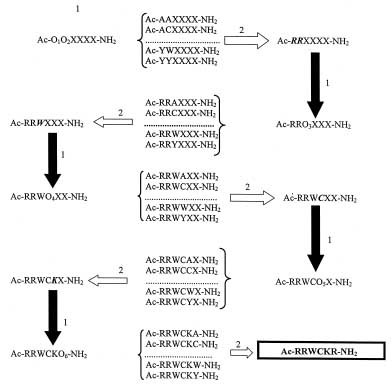
Figure 1. Rastreo de una quimioteca mediante formato iterativo. 1 (flechas negras): síntesis; 2 (flechas blancas): rastreo y selección de las secuencias más activas. Ac: acetil; O: aminoácido definido; X: mezcla equimolar de los 20 L-aminoácidos.
Proceso iterativo
En este proceso se identifica, en cada paso de descodificación, un aminoácido para una posición definida de la secuencia activa. Las primeras quimiotecas que se crearon consistían en secuencias de seis aminoácidos (16, 24). Un ejemplo sería la fórmula Ac-O1O2XXXX-NH2, donde O1O2 son las dos primeras posiciones que se definen con un aminoácido determinado: AA, AC, AD... hasta VY, WY, YY (utilizando el código de una letra para cada aminoácido), para un total de 400 combinaciones (202). En cada una de estas 400 combinaciones las X hacen referencia a una mezcla equimolar de 19 de los 20 L-aminoácidos naturales (la cisteína se omite en las X, pero no así en las posiciones definidas, representadas como O), lo que hace un total de 130.321 posibles combinaciones (194). Así, cada una de las 400 mezclas se compone de 130.321 (194) péptidos y, en total, en la quimioteca tendríamos 52.321.400 (400 x 194) péptidos. Cada una de estas 400 mezclas se somete, por separado, al ensayo biológico de interés para identificar los aminoácidos más activos en las posiciones O1O2, que corresponden a RR en el ejemplo de la Fig. 1. Seguidamente se procede a la síntesis de ya sólo 20 nuevas mezclas, Ac-RRO3XXX-NH2, y al ensayo e identificación del aminoácido más activo en la posición O3, que resulta ser W (Fig. 1); y así sucesivamente hasta encontrar las secuencia más activa, que en este caso, a modo de ejemplo, sería Ac-RRWCKR-NH2.
La naturaleza de la secuencia activa viene definida por el ensayo biológico, es decir, con la misma quimioteca podemos encontrar otras secuencias activas para diferentes ensayos biológicos que se planteen. Utilizando esta aproximación iterativa se requieren de 6 a 12 semanas para obtener secuencias peptídicas activas.
Aunque era un método novedoso y relativamente rápido (puesto que se ensayan más de 50 millones de moléculas diferentes), el ensayo de 400 muestras todavía parecía optimizable en tiempo y por ello se idearon las quimiotecas combinatorias en formato de rastreo posicional.
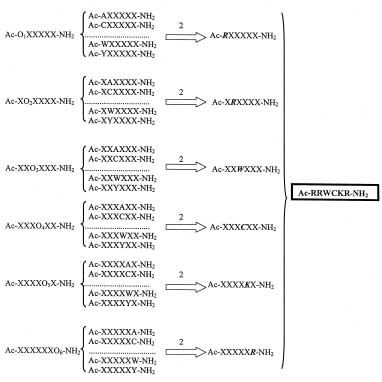
Figure 2. Rastreo de una quimioteca mediante formato de rastreo posicional. 2 (flechas blancas): rastreo y selección de las secuencias más activas. Ac: acetil; O: aminoácido definido; X: mezcla equimolar de los 20 L-aminoácidos.
Rastreo posicional
El uso del procedimiento de rastreo posicional no requiere la repetición de los pasos de ensayo, selección y síntesis, sino que en un solo ensayo y en una sola semana, en función de la bondad del ensayo, se pueden identificar secuencias peptídicas con actividad biológica.
En las quimiotecas combinatorias en formato de rastreo posicional lo que se trata es de sintetizar tantas subquimiotecas como aminoácidos contenga la secuencia y, en cada una de ellas, definir una posición, y las restantes son mezclas equimolares de los aminoácidos (26). Por ejemplo, en el caso de una secuencia de seis aminoácidos tendremos seis subquimiotecas posicionales diferentes, cada una de las cuales tendría la siguiente forma: Ac-O1XXXXX-NH2, Ac-XO2 XXXX-NH2, Ac-XXO3XXX-NH2, Ac-XXXO4XX-NH2, Ac- XXXXO5X-NH2, Ac-XXXXXO6-NH2 (donde O1, O2... O6 vendrían ocupadas por los 20 aminoácidos) (Fig. 2). En total hay 120 mezclas (6 posiciones definidas � 20 aminoácidos posibles para ocuparlas) y cada una de ellas cuenta con 2,5 millones de combinaciones posibles (195); así, cada una de las seis subquimiotecas contiene unos 50 millones (20 x 195) de hexámeros diferentes.
La ventaja de las quimiotecas en formato de rastreo posicional reside en que cada una de las posiciones se ensaya independientemente del resto, por lo que ya no se precisan los procesos iterativos de selección y síntesis requeridos en las quimiotecas anteriormente descritas. En la Fig. 2 se muestra un ejemplo de una quimioteca en formato de rastreo posicional. Una vez sintetizada la quimioteca se realiza el ensayo biológico y obtendríamos, a partir de la lectura de los resultados, los aminoácidos más activos en cada una de las posiciones. Únicamente restaría sintetizar las moléculas de secuencia totalmente definida y ensayarlas de nuevo para validar la quimioteca y comprobar que tienen las características biológicas exigidas por el ensayo.
DESARROLLO DE NUEVOS ANTIMICROBIANOS UTILIZANDO QUIMIOTECAS SOLUBLES
Quimiotecas de L-aminoácidos
Las quimiotecas de péptidos fueron las primeras en sintetizarse, debido a que los métodos de síntesis peptídica son los mejor desarrollados. Además, aunque tradicionalmente no se han utilizado como agentes terapéuticos por su baja biodisponibilidad oral y su alta degradabilidad enzimática, tanto los insectos como los mamíferos y las plantas liberan péptidos de bajo peso molecular como uno de sus primeros sistemas defensivos para luchar contra las infecciones bacterianas (27). Debido a que poseen un mecanismo de acción diferente al de los antibióticos clásicos (los péptidos generalmente ejercen su acción alterando las propiedades de permeabilidad de las membranas bacterianas) (28), los péptidos pueden desempeñar un importante papel como nuevos agentes en la lucha contra las cepas bacterianas resistentes que van apareciendo y, de hecho, hay péptidos que se vienen utilizando como antibióticos, por ejemplo la polimixina y la bacitracina (29, 30). De ahí que varias de estas quimiotecas se hayan utilizado para la identificación de compuestos con actividad antimicrobiana.
El laboratorio liderado por Richard Houghten (Torrey Pines Institute for Molecular Studies, San Diego, California, EE.UU.) fue el primero en utilizar las quimiotecas solubles para este fin. A partir de una quimioteca de hexapéptidos encontraron secuencias peptídicas que tenían una alta actividad bactericida a concentración micromolar frente a bacterias grampositivas y gramnegativas y frente a la levadura Candida albicans, mientras que eran inocuas para los eritrocitos (16, 24, 31). Sus actividades antimicrobianas frente a cepas resistentes de S. aureus, E. coli, P. aeruginosa y C. albicans las podemos ver en la Tabla 1. Aunque estos péptidos presentan la mitad de actividad que los antibióticos comerciales existentes, sus actividades son del orden, o incluso mayores, que las de otros péptidos antimicrobianos de origen natural, como la cecropina o la magainina.
Otra estrategia es, a partir de péptidos con actividad antimicrobiana conocida y mediante síntesis química combinatoria, introducir en algunas posiciones mezclas equimolares de los 20 L-aminoácidos con el fin de encontrar nuevas secuencias con una mayor actividad; a esto se le llama "combinatorializar" las posiciones. Este tipo de quimiotecas se denominan quimiotecas definidas conformacionalmente, debido a que la estructura de todos los péptidos que componen la quimioteca deriva de un péptido con estructura y conformación definidas. Esta "combinatorialización" no sólo permite el descubrimiento de nuevos análogos más activos en comparación con la secuencia original, sino que posibilita realizar valiosos estudios relacionados con la estructura y la actividad de los péptidos. Blondelle y cols. (32) utilizaron esta estrategia y, basándose en el péptido Ac-LKLLKKLLKKLKKLLKKL-NH2, de conocida actividad antimicrobiana, sintetizaron dos quimiotecas definidas conformacionalmente para tratar de identificar péptidos con actividad antimicrobiana. Después de rastrear las quimiotecas se identificaron algunas secuencias que presentaban una mayor actividad que la original.
Recientemente Hong y cols. (33), utilizando la misma estrategia, han identificado decapéptidos con actividad antimicrobiana frente a varias bacterias, entre ellas Staphylococcus epidermidis, P. aeruginosa, E. coli y la levadura C. albicans, y que presentan baja actividad hemolítica.
Péptidos con D-aminoácidos y con aminoácidos no naturales
Como se ha comentado anteriormente, el principal problema de los péptidos constituidos por L-aminoácidos naturales es su baja estabilidad (por ejemplo frente a las proteasas), con lo que se dificulta su utilización como agentes terapéuticos. Una forma de prolongar la vida media de los péptidos antibacterianos en el organismo es incorporar en las quimiotecas D-aminoácidos u otros aminoácidos no naturales que, al no ser reconocidos por las proteasas, son mucho más resistentes a su degradación y resultan más interesantes como agentes terapéuticos.
En un primer intento se sintetizó una quimioteca de tetrapéptidos a partir de mezclas de aminoácidos L, D o no naturales (34). La primera posición de la quimioteca consistía en un aminoácido definido, que podía ser uno de los 58 aminoácidos diferentes de que disponían los autores (posición U). Las tres posiciones restantes (Z) consistían en mezclas equimolares de 56 de los mismos aminoácidos (L- y D-cisteína se omitieron). Por tanto, la quimioteca era de la forma Ac-UZZZ-NH2. Se obtuvieron así 58 subquimiotecas con 175.616 (562) tetrapéptidos individuales cada una, en total 10.185.728 tetrapéptidos diferentes. Después del rastreo mediante el método iterativo se obtuvo una serie de compuestos con una gran actividad antimicrobiana. A diferencia de los péptidos comentados anteriormente, estos antibióticos parecen ser bacteriostáticos y además tienen una actividad hemolítica menor que los péptidos con L-aminoácidos.
Compuestos peptidomiméticosy moléculas orgánicas
Con el fin de aumentar el repertorio de la diversidad química más allá del uso de aminoácidos no proteogénicos, se han desarrollado quimiotecas de peptidomiméticos o de pequeñas moléculas orgánicas. A los inconvenientes que presentan los péptidos como fármacos, ya comentados, tenemos que añadir su baja selectividad de acción, resultado de la alta flexibilidad conformacional que poseen, lo que posibilita la unión a varios receptores y, en algunos casos, la aparición de reacciones inmunitarias descritas tras la administración de fármacos peptídicos. Con el fin de subsanar estos inconvenientes y a la vez aprovechar el amplio potencial que ofrecen los péptidos para el desarrollo de nuevos compuestos bioactivos, surge el concepto de "peptidomimético" para definir aquellas estructuras no peptídicas capaces de mimetizar o bloquear el efecto biológico de algunos péptidos. Un ejemplo de un peptidomimético sería la morfina, que con una estructura muy alejada de la peptídica es capaz de unirse a receptores peptídicos y mimetizar la acción de péptidos naturales como son las encefalinas. Se espera que estos compuestos sean mejores como medicamentos orales que los péptidos (por las características ya mencionadas).
Actualmente, con el desarrollo de las quimiotecas combinatorias, la búsqueda de compuestos peptidomiméticos se ha incrementado enormemente. Sin embargo, el desarrollo de quimiotecas de moléculas orgánicas no ha sido posible hasta fechas muy recientes debido a que la química orgánica en fase sólida, verdadero motor de las técnicas combinatorias, aún está en pleno crecimiento. Así, no fue hasta 1992 cuando Bunin y Ellman describieron la primera quimioteca combinatoria en química orgánica: una quimioteca de 1,4-benzodiacepinas (35). Hasta ese año el campo de la química combinatoria había sido de dominio exclusivo de péptidos y oligonucleótidos. A partir de ese momento la síntesis de quimiotecas orgánicas ha crecido casi exponencialmente gracias al desarrollo de nuevas resinas, reactivos de acoplamiento, puesta a punto de reacciones, etc. Así, por ejemplo, se han descrito quimiotecas combinatorias de poliaminas, poliglicinas (36), oligocarbamatos (37), hidantoínas (38), ß-mercaptocetonas (39), derivados de cubanos (40) y triazinas (41). Hasta ahora sólo la quimioteca de triazinas se ha ensayado en la identificación de compuestos con actividad antibiótica. Los autores sintetizaron una quimioteca de triazinas con 46.000 (363) compuestos diferentes, y aunque las actividades obtenidas aún están lejos de los antibióticos comerciales (una CMI de 4 µg/ml para Bacteroides subtilis y S. aureus) se pone de manifiesto que el uso de quimiotecas orgánicas puede ser una muy buena vía para el desarrollo de antibióticos.
CONCLUSIONES
La aparición de cepas de bacterias resistentes a los antibióticos en uso, mediante selección natural o forzada por su empleo incontrolado, se plantea como un problema de interés sanitario general. Las nuevas y revolucionarias tecnologías de síntesis química y la automatización de los ensayos están poniendo millones de moléculas a disposición de los procesos de identificación de nuevos antibióticos.
La aplicación de la química combinatoria al desarrollo de fármacos permitirá el descubrimiento de compuestos químicos que contribuirán a la identificación de nuevos fármacos cabeza de serie. Será después de la optimización farmacológica de los mismos cuando estemos en condiciones de proporcionar a la comunidad médica en general nuevas moléculas que, con mucho, sean más eficaces en el tratamiento de las infecciones; moléculas que podrían escapar a los mecanismos enzimáticos de defensa que posee el huésped y que, por lo tanto, ejercerían su función sin ningún tipo de interferencia, o moléculas que mejoraran las propiedades antibióticas de algunas ya existentes.
BIBLIOGRAFÍA