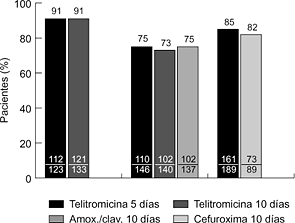
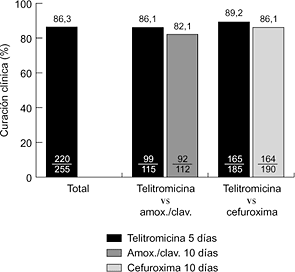
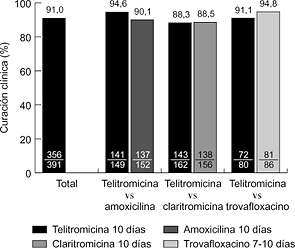
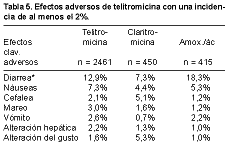
2006 Nueva Estrategias Antiinfecciosas: Interacciones Vacunas-Antimicrobiano
Nuevas estrategias antiinfecciosas: interacciones vacunas-antibióticos
Ponencias: ¿Afecta la vacunación a la prescripción de antibióticos? [pdf]
J. CamposNuevas vacunas antineumocócicas. Más allá del neumococo [pdf]
J. García SiciliaCambios clínicos y terapéuticos de las otitis con la vacunación antineumocócica [pdf]
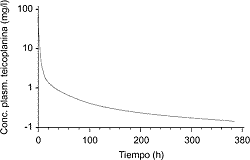
F. Baquero Jr.Inmunización y antimicrobianos. ¿Asociación de efectos frente a Streptococcus pneumoniae? [pdf]
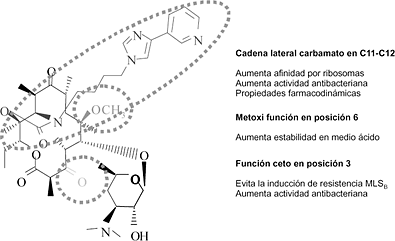
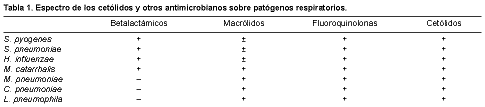
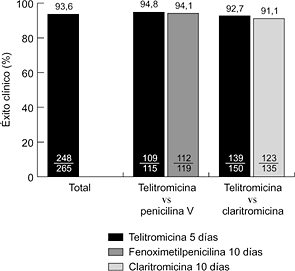
L. AguilarRepercusión de la vacuna contra el neumococo en las resistencias [pdf]
J. J. Granizo.Criterios de introducción de las vacunas en los calendarios vacunales [pdf]
R. RamírezConsideraciones sobre la infección neumocócica pediátrica en Navarra [pdf]
E. BernaolaConclusiones [pdf]
J. J. Picazo