2001 Nuevos antimicrobianos frente a microorganismos grampositivos
- Aspectos microbiológicos de los glucopéptidos y las oxazolidinonas
- Farmacocinética comparada de glucopéptidos y oxazolidinonas
- Aportaciones microbiológicas de los cetólidos
- Mecanismo de acción de los cetólidos y aparición de resistencias
- Eficacia terapéutica de los cetólidos
- Conducta terapéutica ante infecciones hospitalarias y de la comunidad por grampositivos
Aspectos microbiológicos de los glucopéptidos y las oxazolidinonas
C. Betriu Cabeceran
Servicio de Microbiología Clínica, Hospital Clínico San Carlos, Madrid
Mecanismo De Acción De Los Glucopéptidos
Los glucopéptidos son un grupo de antimicrobianos con estructura peptídica que presentan un espectro de actividad similar, restringido a las bacterias grampositivas aerobias y anaerobias. En las dos últimas décadas han adquirido una mayor importancia en el tratamiento de las infecciones causadas por microorganismos grampositivos multirresistentes. La vancomicina y la teicoplanina son los únicos antibióticos de este grupo actualmente disponibles; otros glucopéptidos en estudio son la decaplanina, LY 333328 (oritavancina), MDL 62208, MDL 62211, MDL 62879 y Bi 397.
La vancomicina, producida por Streptomyces orientalis, se introdujo en el mercado en 1958. Sin embargo, debido a su toxicidad y a la aparición de penicilinas resistentes a las penicilinasas, se utilizó poco. Posteriormente, el considerable aumento de la resistencia a la meticilina de Staphylococcus aureus y a los betalactámicos en otros grampositivos, así como la obtención de preparados más purificados con la consiguiente disminución de efectos indeseables, han hecho que su utilización haya aumentado.
La teicoplanina, producida por Actynoplanes teichomycetus, en un principio se denominó teicomicina A2 y presenta una estructura semejante a la de la vancomicina. Su mecanismo de acción consiste en inhibir la síntesis de la pared bacteriana, impidiendo la polimerización del peptidoglicano de la pared celular. Concretamente, los glucopéptidos se unen al terminal D-alanil-d-alanina del pentapéptido precursor del peptidoglicano mediante cinco puentes de hidrógeno. Secundariamente, al inhibir el crecimiento celular alteran la permeabilidad celular y la síntesis de RNA. Son bactericidas frente a la mayoría de los microorganismos grampositivos, exceptuando los enterococos. Actúan solamente sobre microorganismos en crecimiento activo. Presentan actividad frente a los microorganismos grampositivos aerobios y anaerobios (estafilococos, estreptococos, Corynebacterium diphteriae, Listeria monocytogenes, cocos anaerobios, Clostridium spp., Eubacterium spp. y Actynomyces spp.).
Resistencias A Los Glucopéptidos
Los primeros aislamientos de enterococos resistentes a la vancomicina se describieron en Inglaterra en 1988, y desde entonces se han aislado en numerosos países. En la actualidad, su incidencia varía según la localización geográfica. En Estados Unidos es donde hay una mayor proporción de enterococos resistentes a la vancomicina. Según los datos del estudio internacional SENTRY (1), correspondiente al periodo 1997-1999, en el que se incluyeron casi 5000 aislamientos de enterococos, el porcentaje de aislamientos resistentes a la vancomicina en Estados Unidos en 1999 fue del 17%, mientras que en Europa y en otras áreas geográficas las tasas de resistencia a la vancomicina fueron muy pequeñas (entre el 1% y el 3%), y no cambiaron significativamente a lo largo del periodo estudiado.
La resistencia de los enterococos a los glucopéptidos se ha clasificado en diferentes fenotipos; hasta el momento actual se han descrito seis: VanA, VanB, VanC, VanD, VanE y VanG.
Los estafilococos coagulasa negativos (Staphylococcus haemolyticus y Staphylococcus epidermidis, principalmente) resistentes a los glucopéptidos se empezaron a describir en 1981, siendo más común la resistencia a la teicoplanina que a la vancomicina. Posteriormente se han comunicado, entre los S. aureus resistentes a la meticilina (SARM), los siguientes tipos de aislamientos:
– Cepas GISA: S. aureus resistentes a la meticilina y con sensibilidad disminuida a la vancomicina (CMI 8-16 mg/l) o a la teicoplanina (CMI 16 mg/l). El primer aislamiento se describió en Japón en 1997 (2) y posteriormente se han comunicado otros aislamientos con las mismas características en Estados Unidos, Francia, Hong-Kong y Corea (3-8).
– Cepas heteroGISA: S. aureus sensibles a la vancomicina pero que contienen subpoblaciones (frecuencia 1 ´ 10-6 colonias) que crecen en presencia de vancomicina >4 mg/l. Se han aislado cepas heterorresistentes a los glucopéptidos en zonas geográficas donde la prevalencia de SARM es alta. Sin embargo, la incidencia de h-GISA es muy variable (9-11). El significado clínico de estas cepas no está claro; se ha sugerido que los h-GISA podrían ser precursores de GISA y pueden estar, en algunos casos, asociados con fracasos terapéuticos.
La disminución de la sensibilidad a la vancomicina en SARM no parece deberse a la transferencia de genes de los enterococos resistentes a la vancomicina, sino que podría estar producida por un proceso de selección gradual debido a la presión selectiva ejercida por el antibiótico.
En un modelo experimental de meningitis por una cepa de Streptococcus pneumoniae tolerante a la vancomicina se produjo fracaso terapéutico (12). Recientemente se ha comunicado un caso de fracaso clínico con vancomicina en un paciente con meningitis del cual se aisló una cepa de neumococo tolerante a la vancomicina (13). En un estudio llevado a cabo en Suecia (14) se detectaron tres cepas tolerantes a la vancomicina de un total de 116 neumococos. En España no se ha encontrado, por ahora, ninguna cepa de neumococo con estas características (15).
A lo largo de los últimos 15 años ha aumentado la incidencia de infecciones por microorganismos grampositivos multirresistentes, entre los que se incluyen S. pneumoniae resistente a la penicilina y que presenta, además, resistencia a cefalosporinas, macrólidos, tetraciclinas y cotrimoxazol; S. aureus resistente a meticilina; S. aureus con sensibilidad disminuida a la vancomicina o con heterorresistencia a la vancomicina; estafilococos coagulasa negativos resistentes a los glucopéptidos y enterococos resistentes a los glucopéptidos. La aparición y el aumento de estas resistencias ha dado lugar a una mayor preocupación por el uso adecuado de los antibióticos y ha creado la necesidad de contar con nuevos fármacos.
Mecanismo De Accioón De Las Oxazolidinonas
Descubiertas en 1987, las oxazolidinonas son quimioterápicos obtenidos por síntesis que comparten una estructura básica bicíclica, la feniloxazolidin-2-ona. Linezolid (PNU-100766) y eperezolid (PNU-100592), los dos primeros miembros de esta familia, son muy similares. Presentan biodisponibilidad por vía oral y parenteral. Son bacteriostáticos frente a estafilococos y enterococos, y pueden ser bactericidas frente a algunos estreptococos, incluyendo S. pneumoniae.
Actúan inhibiendo la síntesis de proteínas. Se unen a la subunidad 50S e impiden la formación del complejo de iniciación entre la subunidad 30S o 70S, el RNAm, los factores de iniciación IF2 e IF3, y la f-Met-RNAt. Estos compuestos son únicos, actúan al principio de la traducción. Aunque el cloranfenicol y la lincomicina compiten por el punto de unión ribosómico, se ha visto que el mecanismo de las oxazolidinonas es diferente. El cloranfenicol y la lincomicina actúan sobre la elongación (actividad peptidiltransferasa) y la terminación de la traducción, mientras que las oxazolidinonas no intervienen en estas fases.
El linezolid ha sido aprobado recientemente para el tratamiento de las neumonías e infecciones de piel y estructuras cutáneas. Su espectro de actividad incluye a estreptococos, enterococos, estafilococos, bacilos grampositivos como L. monocytogenes, Corynebacterium spp., Bacillus spp., Nocardia spp., Rhodococcus spp., Mycobacterium tuberculosis, cocos grampositivos anaerobios y especies de Clostridium. Presenta cierta actividad frente a algunas bacterias gramnegativas aerobias, como Haemophilus influenzae, Moraxella catarrhalis, Bordetella pertussis, Legionella spp. y Neisseria gonorrhoeae, y sobre algunos bacilos gramnegativos anaerobios (Bacteroides spp., Prevotella spp. y Fusobacterium spp.). El NCCLS todavía no ha establecido los puntos de corte para el linezolid. De acuerdo con las características farmacocinéticas, se consideran las cepas sensibles cuando la CMI es <4 mg/l.
El linezolid es activo frente a microorganismos grampositivos resistentes a otros antimicrobianos. Presenta una buena actividad frente a SARM, comparable a la de la vancomicina y la teicopianina, y no se ha detectado resistencia cruzada con otros antibióticos (16-19). Se ha comprobado también la excelente actividad del linezolid frente a enterococos, independientemente de su sensibilidad a la vancomicina o del fenotipo de resistencia (CMI50 1-2 mg/l y CMI90 2-4 mg/l) (17, 20).
El linezolid presenta similar actividad frente a los neumococos sensibles a la penicilina y frente a los que tienen resistencia intermedia o son resistentes (17, 20). A una concentración <2 mg/l el linezolid inhibe el crecimiento de los estreptococos (Streptococcus pyogenes, Streptococcus agalactiae y S. pneumoniae) resistentes a la eritromicina (19), lo que hace que este nuevo antimicrobiano pueda ser considerado una alternativa válida para el tratamiento de las infecciones ocasionadas por estreptococos resistentes a la eritromicina.
En estudios realizados con bacterias anaerobias (21, 22), el linezolid ha mostrado una mayor actividad frente a Fusobacterium spp., y los valores más altos de CMI50 se observaron frente a Bacteroides fragilis, especies de Prevotella y Clostridium difficile.
En un estudio llevado a cabo con estreptococos de los grupos C y G, en el cual el 75% de las cepas eran tolerantes a la vancomicina, el linezolid mostró buena actividad frente a todos los estreptococos estudiados, con unos valores de CMI50 y CMI90 de 2 mg/l (23). Dada la excelente actividad mostrada por el linezolid, este antibiótico podría ser considerado como monoterapia o en asociación con otros agentes en el tratamiento de las infecciones invasoras por estreptococos de los grupos C y G en pacientes de alto riesgo y que no puedan ser tratados con penicilina.
El linezolid ha mostrado una ligera actividad frente a M. catarrhalis (CMI50 4 mg/l, CMI90 16 mg/l) y limitada actividad frente a H. influenzae (CMI50 16 mg/l, CMI90 32 mg/l) (17).
La presencia de genes que confieren resistencia a los antimicrobianos que inhiben la síntesis de proteínas no influye en la actividad in vitro del linezolid frente a los cocos grampositivos. Se ha comprobado que el linezolid es activo frente a Enterococcus faecalis y S. aureus resistentes a los macrólidos, estreptograminas, tetraciclinas, cloranfenicol o aminoglucósidos (24).
La resistencia in vitro no se ha podido inducir fácilmente en bacterias grampositivas como S. aureus y S. epidermidis tras la exposición a concentraciones de linezolid de 2, 4 y 8 veces la CMI (25). Sin embargo, aunque es difícil que se produzcan resistencias, se ha comunicado la aparición de resistencia a linezolid en dos pacientes durante el protocolo de uso compasivo (26). Los pacientes recibieron 4 o 6 semanas de tratamiento para una infección por Enterococcus faecium de evolución clínica complicada. Presentaron bacteriemia y eran portadores de catéteres permanentes desde hacía mucho tiempo y que no se podían retirar. En abril de 2001, Gonzales y cols. (27) describieron cinco casos de pacientes con infección por enterococos resistentes a la vancomicina resistentes al linezolid; todos habían recibido tratamiento prolongado con este antibiótico (21 a 40 días). Posteriormente se ha comunicado el primer aislamiento de SARM resistente al linezolid (28).
Bibliografía
1. Low, D.E., Keller, N., Barth, A., Jones, R.N. Clinical prevalence, antimicrobial susceptibility and geographic resistance patterns of enterococci: Results from the SENTRY Antimicrobial Surveillance Program, 1997-1999. Clin Infect Dis 2001; 32 (Suppl. 2): S133-S145.
2. Hiramatsu, K., Hanaki, H., Ino, T., Yabuta, K., Oguri, T., Tenover, F.C. Methicillin-resistant Staphylococcus aureus clinical strain with reduced vancomycin susceptibility. J Antimicrob Chemother 1997; 40: 135-136.
3. Smith, T.L., Pearson, M.L., Wilcox, K.R. y cols. Emergence of vancomycin resistance in Staphylococcus aureus. Glycopeptide-intermediate Staphylococcus aureus Working Group. N Engl J Med 1999; 340: 493-501.
4. Sieradzki, K., Roberts, R.B., Haber, S.W., Tomasz, A. The development of vancomycin resistance in a patient with methicillin-resistant Staphylococcus aureus infection. N Engl J Med 1999; 340: 517-523.
5. Fridkin, S.K. Vancomycin-intermediate and -resistant Staphylococcus aureus: What the infectious disease specialist needs to know. Clin Infect Dis 2001; 32: 108-115.
6. Ploy, M.C., Grelaud, C., Martin, C., de Lumley, L., Denis, F. First clinical isolate of vancomycin-intermediate Staphylococcus aureus in a French hospital. Lancet 1998; 351: 1212.
7. McManus, J. Vancomycin resistant Staphylococcus reported in Hong-Kong. BMJ 1999; 318: 626.
8. Mi-Na, K., Pai, C.H., Woo, J.H., Ryu, J.S., Hiramatsu, K. Vancomycin intermediate Staphylococcus aureus in Korea. J Clin Microbiol 2000; 38: 3879-3881.
9. Hiramatsu, K., Aritaka, N., Hanaki, H. y cols. Dissemination in Japanese hospitals of strains of Staphylococcus aureus heterogeneously resistant to vancomycin. Lancet 1997; 350: 1670-1673.
10. Ariza, J., Pujol, M., Cabo, J. y cols. Vancomycin in surgical infections due to methicillin-resistant Staphylococcus aureus with heterogeneous resistance to vancomycin. Lancet 1999; 353: 1587-1588.
11. Marchese, A., Balistreri, G., Tonoli, E., Debbia, E.A., Schito, G.C. Heterogeneous vancomycin resistance in methicillin-resistant Staphylococcus aureus strains isolated in a large Italian hospital. J Clin Microbiol 2000; 38: 866-869.
12. Novak, R., Henriques, B., Charpentier, E., Normark, S., Tuomanen, E. Emergence of vancomycin tolerance in Streptococcus pneumoniae. Nature 1999; 399: 590-593.
13. McCullers, J.A., English, B.K., Novak, R. Isolation and characterization of vancomycin-tolerant Streptococcus pneumoniae from the cerebrospinal fluid of a patient who developed recrudescent meningitis. J Infect Dis 2000; 181: 369-373.
14. Henriques Normark, B., Novak, R., Ortqvist, A., Kallenius, G., Tuomanen, E., Normark, S. Clinical isolates of Streptococcus pneumoniae that exhibit tolerance of vancomycin. Clin Infect Dis 2001; 32: 552-558.
15. Antón, R., Bláquez, R., Gómez-Garcés, J.L., Alós, J.L. Study of vancomycin tolerance in 120 strains of Streptococcus pneumoniae isolated in 1999 in Madrid, Spain. J Antimicrob Chemother 2001; 47: 902-903.
16. Johnson, A.P., Warner, M., Livermore, D.M. Activity of linezolid against multi-resistant grampositive bacteria from diverse hospitals in the United Kingdom. J Antimicrob Chemother 2000; 45: 225-230.
17. Gemmell, C.G. Susceptibility of a variety of clinical isolates to linezolid: A European inter-country comparison. J Antimicrob Chemother 2001; 48: 47-52.
18. von Eiff, C., Peters, G. Comparative in-vitro activities of moxifloxacin, trovafloxacin, quinupristin/dalfopristin and linezolid against staphylococci. J Antimicrob Chemother 1999; 43: 569-573.
19. Betriu, C., Redondo, M., Palau, M.L. y cols. Comparative in vitro activities of linezolid, quinupristin-dalfopristin, moxifloxacin, and trovafloxacin against erythromycin-susceptible and -resistant streptococci. Antimicrob Agents Chemother 2000; 44: 1838-1841.
20. Cercenado, E., García-Garrote, F., Bouza, E. In vitro activity of linezolid against multiply resistant Gram-positive clinical isolates. J Antimicrob Chemother 2001; 47: 77-81.
21. Goldstein, E.J., Citron, D.M., Merriam, C.V. Linezolid activity compared to those of selected macrolides and other agents against aerobic and anaerobic pathogens isolated from soft tissue bite infections in humans. Antimicrob Agents Chemother 1999; 43: 1469-1474.
22. Wise, R., Andrews, J.M., Boswell, F.J., Ashby, J.P. The in-vitro activity of linezolid (U-100766) and tentative breakpoints. J Antimicrob Chemother 1998; 42: 721-728.
23. Zaoutis, T., Moore, L.S., Furness, K., Klein, J.D. In vitro activities of linezolid, meropenem, and quinupristin-dalfopristin against group C and G streptococci, including vancomycin-tolerant isolates. Antimicrob Agents Chemother 2001; 45: 1952-1954.
24. Fines, M., Leclercq, R. Activity of linezolid against Gram-positive cocci possessing genes conferring resistance to protein synthesis inhibitors. J Antimicrob Chemother 2000; 45: 797-802.
25. Zurenko, G.E., Yagi, B.H., Schaadt, R.D. y cols. In vitro activities of U-100592 and U-100766, novel oxazolidinone antibacterial agents. Antimicrob Agents Chemother 1996; 40: 839-845.
26. Zurenko, G.E., Todd, W.M., Hafkin, B.A. Development of linezolid-resistant Enterococcus faecium in two compassionate use program patients treated with linezolid. 39th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC), San Francisco 1999.
27. Gonzales, R.D., Schreckenberger, P.C., Graham, M.B., Kelkar, S., DenBesten, K., Quinn, J.P. Infections due to vancomycin-resistant Enterococcus faecium resistant to linezolid. Lancet 2001; 357: 1179.
28. Tsiodras, S., Gold, H.S., Sakoulas, G. y cols. Linezolid resistance in clinical isolate of Staphylococcus aureus. Lancet 2001; 358: 207-208.
Farmacocinética comparada de glucopéptidos y oxazolidinonas
J.R. Azanza Perea
Servicio de Farmacología Clínica, Clínica Universitaria de Navarra, Universidad de Navarra, Pamplona
Los glucopéptidos y las oxazolidinonas son dos familias de antibacterianos de gran importancia puesto que presentan una actividad notable frente a cocos grampositivos, incluidas las cepas resistentes a otros fármacos. El grupo de los glucopéptidos está formado en la actualidad por dos componentes, vancomicina y teicoplanina, que presentan estructuras químicas muy complicadas, de gran tamaño e importante peso molecular, en las que existen azúcares y estructuras peptídicas, algunas de ellas halogenadas. El linezolid es, al contrario, una estructura química mucho más sencilla y reducida de tamaño perteneciente al grupo de las oxazolidinonas.
De sus estructuras químicas se deduce que van a existir diferencias en el comportamiento general de estos fármacos en cuanto a farmacocinética. La vancomicina y la teicoplanina presentan cierta similitud en alguno de los apartados farmacocinéticos, como puede ser la vía de eliminación o algunas de las características de la distribución, mientras que el linezolid va a tener un perfil distinto.
En la Tabla 1 se describen los parámetros farmacocinéticos obtenidos tras la administración por vía intravascular (intravenosa) de estos tres fármacos. Se aprecia una diferencia importante en el valor de la concentración plasmática máxima (Cmáx), que en el caso de la teicoplanina resulta muy superior a la alcanzada con dosis semejantes de linezolid o vancomicina, incluso aun cuando se corrijan en relación con la dosis administrada. El volumen de distribución es relativamente semejante, lo cual no deja de ser sorprendente si se considera que la fijación de la teicoplanina a las proteínas plasmáticas es muy elevada y claramente superior a la de los otros dos. Además, existen diferencias notables en el aclaramiento, que en el caso de la teicoplanina resulta entre 7 y 10 veces menor que el de la vancomicina y el linezolid, en relación con el mayor tiempo de permanencia en plasma motivado por la fijación a las proteínas. Si se revisan uno a uno los parámetros farmacocinéticos específicos correspondientes a absorción, distribución y eliminación, se pueden explicar las diferencias de interés más práctico.

Absorción
Hay que partir de la consideración de que no es posible administrar la vancomicina por vía intramuscular por su toxicidad para los tejidos. Además, presenta una fracción biodisponible (f) por vía oral inferior al 10%. Ambos hechos suponen que este fármaco sea de uso exclusivo por vía intravenosa, al menos cuando se pretende alcanzar concentraciones plasmáticas óptimas (1, 2). En el extremo opuesto se sitúa el linezolid, que presenta una fracción biodisponible, administrado por vía oral, que puede incluso superar al 100%, y que además tiene una velocidad de absorción elevada, como muestra el valor de la tmáx, situado entre 1 y 2 horas (3, 4). Esta característica permite asegurar la consecución de concentraciones plasmáticas similares con independencia de la vía de administración (oral o intravenosa), lo que a su vez explica la presencia de áreas bajo la curva intravenosa y oral semejantes, siempre que la dosis administrada sea similar. La administración conjunta de linezolid junto con alimentos reduce ligeramente la Cmáx, pero sin que aparentemente tenga repercusión práctica (5).
La teicoplanina se sitúa en un término intermedio, puesto que aunque como la vancomicina es un fármaco con una absorción por vía oral muy deficiente, resulta factible su administración por vía intramuscular y presenta una fracción biodisponible que supera el 90%, con una tmáx de 1 a 3 horas. Al igual que en el caso anterior, esta circunstancia supone que se alcancen concentraciones plasmáticas máximas y áreas bajo la curva muy similares tras la administración intravascular y oral (6, 7).
Probablemente, la mayor repercusión práctica de las diferencias en los parámetros de absorción se concretan en la posibilidad de realizar tratamiento secuencial, vía intravenosa/oral para el linezolid e intravenosa/intramuscular para la teicoplanina, lo que sin duda puede repercutir en la duración del tratamiento hospitalario.
Distribuición
Cualquier lector con cierta formación en farmacocinética ha podido advertir que existe cierta incoherencia en los comentarios realizados al inicio de este breve texto, cuando se señala que el volumen de distribución de la teicoplanina es parecido al de la vancomicina y el linezolid cuando el primero de ellos circula en plasma fijado a proteínas con gran intensidad y, fruto de ello, alcanza una concentración plasmática superior a los otros. La incoherencia puede acrecentarse si se calcula la magnitud del Vd de los tres fármacos basándose en la dosis administrada y la concentración plasmática alcanzada (5, 8, 9). La explicación de este contrasentido tiene su base en el uso, en este caso para teicoplanina, del valor del Vdss, es decir, del calculado cuando se ha alcanzado el equilibrio de distribución. La realidad es que la teicoplanina presenta una curva de concentraciones plasmáticas, incluso cuando se administra en bolo intravenoso, en la que se pueden identificar hasta tres subcurvas (Fig. 1). En la primera de ellas se produce una caída rapidísima de las concentraciones plasmáticas, que no puede justificarse en ningún caso por la eliminación, pues el aclaramiento de este fármaco es muy reducido, y por consiguiente sólo se explica por la rápida difusión del fármaco a los tejidos. Esta primera fase es tan rápida que la semivida de la teicoplanina, calculada de forma específica para esta fase, es de 20 a 30 minutos, lo que de hecho significa que en 30 minutos la concentración plasmática de teicoplanina se reduce a la mitad, porque una parte del fármaco ha sido capaz de difundir a los tejidos a pesar del elevado porcentaje de fijación a las proteínas (9). Tras los primeros minutos la curva de concentraciones cambia de pendiente, se suaviza, y se inicia con ello otra fase que dura más tiempo, 2-3 horas, en las que se puede estimar una semivida de 1,6 a 4 horas. En ésta parecen coexistir dos componentes; por un lado, el fármaco presente en el plasma puede empezar a ser eliminado, y por otro continúa el proceso de distribución. Este último es, en estos momentos, un poco especial, puesto que el fármaco estará pasando desde el compartimiento periférico superficial, al que ha distribuido con gran velocidad, al compartimiento periférico profundo, y la cantidad difundida a éste permitirá la difusión de la teicoplanina desde el plasma al compartimiento periférico en un proceso secuencial en el cual los gradientes de concentraciones adquieren importancia. Lógicamente, este proceso es más lento que el anterior, y de ahí que la curva se suavice. Posteriormente la curva sufre un notable aplanamiento, hasta el punto de que la semivida calculada para esta fase puede superar las 140 horas.
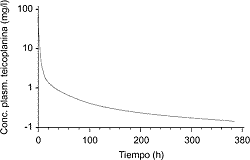
Figura 1.
El valor del Vd va a depender entonces del tiempo elegido para su cálculo. Si se utiliza el mismo sistema que con otro fármaco y se utiliza la Cmáx real, la cifra será muy pequeña, lo que sin duda no expresa la realidad distributiva; en cambio, si se calcula tras la fase de distribución y en el estado de equilibrio, la cifra adquiere un valor representativo. Esta explicación justifica que, en realidad, las concentraciones hísticas alcanzadas por la teicoplanina sean adecuadas a pesar de su elevada fijación proteica, y que únicamente en aquellos tejidos con problemas habituales en la distribución de fármacos, como el tejido adiposo o el LCR, se pueda encontrar una concentración reducida con la dosis habitual. En esta misma situación se sitúa la vancomicina, mientras que la teicoplanina alcanza concentraciones hísticas más elevadas, y especialmente llamativo es el hecho de que entre ellas se encuentre el LCR.
Eliminación
La eliminación de estos fármacos desde el plasma se realiza de forma rápida, especialmente en el caso del linezolid y la vancomicina, y mucho más lentamente la teicoplanina. Precisamente, este aclaramiento reducido, en relación con una fijación a proteínas elevada, es el que justifica que la semivida de eliminación de este fármaco sea diez veces superior a la que alcanzan el linezolid o la vancomicina.
Cuando se estudia el aclaramiento renal de estos fármacos se objetiva que, en el caso de la vancomicina y la teicoplanina, su valor es muy próximo al del aclaramiento total, lo que expresa que ambos fármacos se eliminan casi en su totalidad por la orina en su forma activa. En el caso del linezolid el aclaramiento renal supone cifras próximas o inferiores al 50% del total, por lo que se puede considerar la existencia de metabolismo que se concreta en la oxidación del anillo morfolino con la formación de hasta dos metabolitos inactivos en ambos casos, que se eliminan por la orina y por las heces. Es llamativo que el proceso metabólico del linezolid se realice por oxidación no microsomal, lo cual excluye a este fármaco de la posibilidad de que pueda estar implicado en interacciones por participación de las diferentes isoenzimas del CYP450 (10, 11).
Es bien conocido que la insuficiencia renal modifica de forma muy importante la cinética de eliminación de la vancomicina, hasta el punto de que, en relación con la toxicidad de este fármaco, se recomiende el ajuste posológico en los pacientes que presentan esta anomalía, según la monitorización de las concentraciones plasmáticas, con el fin de evitar la toxicidad dependiente de la concentración (12). En el caso de la teicoplanina se recomienda ajustar la dosis cuando el aclaramiento de creatinina es inferior a 25 ml/min (13), mientras que con el linezolid no se precisa ajustar la posología en caso de insuficiencia renal (14).
Bibliografía
1. Tedesco, F., Markham, R., Gurwith, M., Christie, D., Bartlett, J.G. Oral vancomycin for antibiotic-associated pseudomembranous colitis. Lancet 1978; ii: 226-228.
2. Marrie, T.J., Faulkner, R.S., Badley, B.W.D., Hartlen, M.R., Comeau, S.A., Miller, H.R. Pseudomembranous colitis: Isolation of two species of cytotoxic clostridia and successful treatment with vancomycin. Can Med Assoc J 1978; 119: 1058-1060.
3. Stalker, D.J., Wajszczuk, C.P., Batts, D.H. Linezolid safety, tolerance, and pharmacokinetics following oral dosing twice daily for 14,5 days. 37th Interscience Conference on Antimicrobial Agents and Chemotherapy, Toronto 1997; 23.
4. Welshman, I.R., Stalker, D.J., Wajszczuk, C.P. Assessment of absolute bioavailability and evaluation of the effect of food on oral bioavailability of linezolid. Antiinfect Drugs Chemother 1998; 1 (Suppl. 16): 54.
5. Pawsey, S.D., Daley-Yates, P.T., Wajszczuk, C.P. U-100766 safety, toleration and pharmacokinetics after oral and intravenous administration. 1st European Congress on Chemotherapy, Glasgow 1996; F151.
6. Maskell, J.P., Carter, J.L.B., Boyd, R.B., Williams, R.J. Teicoplanin as a prophylactic antibiotic for dental bacteraemia. J Antimicrob Chemother 1986; 17: 651-659.
7. Ripa, S., Ferrante, L., Mignini, F., Falcioni, E. Pharmacokinetics of teicoplanin. Chemotherapy 1988; 34: 178-184.
8. Moellering, R.C. Pharmacokinetics of vancomycin. J Antimicrob Chemother 1984; 14: 43-52.
9. Rowland, M. Clinical pharmacokinetics of teicoplanin. Clin Pharmacokinet 1990; 18: 184-209.
10. Wienkers, L.C., Wynalda, M.A., Feenstra, K.L. y cols. In vitro metabolism of linezolid (PNU-100766): Lack of induction or inhibition of cytochrome P450 enzymes and studies on the mechanism of formation of the major human metabolite, PNU-142586. 39th Interscience Conference on Antimicrobial Agents and Chemotherapy, San Francisco 1999; A68.
11. Feenstra, K.L., Slatter, J.G., Stalker, D.J. y cols. Metabolism and excretion of the oxazolidinone antibiotic linezolid (PNU-100766) following oral administration of [14C]PNU 100766 to healthy human volunteers. 38th Interscience Conference on Antimicrobial Agents and Chemotherapy, San Diego 1998; 17.
12. Moellering, R.C., Krogstad, D.J. Jr., Greenblatt, D.J. Pharmacokinetics of vancomycin in normal subjects and in patients with reduced renal function. Rev Infect Dis 1981; 3: S230-S235.
13. Bonati, M., Traina, G.L., Rosina, R., Buniva, G. Pharmacokinetics of a single intravenous dose of teicoplanin in subjects with various degrees of renal impairment. J Antimicrob Chemother 1988; 21 (Suppl. A): 29-37.
14. Brier, M.E., Stalker, D.J., Aronoff, G.R. y cols. Pharmacokinetics of linezolid in subjects with varying degrees of renal function and on dialysis. J Invest Med 1998; 46: 276A.
Mecanismo de acción de los cetólidos y aparición de resistencias
C. Gimeno Cardona
Servicio y Departamento de Microbiología, Hospital Clínico y Facultad de Medicina de Valencia
Los antibióticos macrólidos se han utilizado desde mediados del siglo pasado para el tratamiento de distintos tipos de procesos infecciosos, especialmente las infecciones de vías respiratorias, que en la actualidad siguen siendo una causa importante de mortalidad al originar alrededor de 50 millones de muertes al año, según la OMS.
Como ha venido ocurriendo en general en el campo de la quimioterapia, diferentes son los motivos que hacen necesaria la búsqueda de nuevos agentes con el objeto de mejorar el perfil de los ya existentes. Entre estas razones cabe destacar la necesidad de compuestos con una actividad antimicrobiana superior, de más amplio espectro, capaces de vencer eficazmente las resistencias y con una mejor farmacología.
Los cetólidos constituyen un nuevo grupo de antimicrobianos, derivados de los macrólidos, aparecidos por primera vez en la década de 1990 en congresos y reuniones de quimioterapia (1). Su denominación proviene de la sustitución de un grupo ceto en posición 3 del anillo macrólido en lugar del azúcar cladinosa presente en la eritromicina, el antibiótico tipo del grupo. Numerosos han sido los compuestos diseñados hasta ahora, de los cuales los más activos son los que poseen en posición C11-C12 un grupo carbamato (carbamato cetólidos). Dentro de este grupo, dos son los antimicrobianos más desarrollados: telitromicina y ABT-773 (2). El mecanismo de acción es, igual que en los macrólidos, la inhibición de la síntesis proteica en la subunidad 50s de los ribosomas bacterianos, si bien existen algunas diferencias en la interacción con los ribosomas.
Las aportaciones microbiológicas de los cetólidos pueden ser estudiadas desde tres puntos de vista: actividad in vitro, capacidad bactericida y estudio del efecto postantibiótico, y por último características farmacocinéticas favorecedoras de la acción frente a los microorganismos.
Actividad In Vitro
Microorganismos grampositivos
Streptococcus pneumoniae
Los resultados obtenidos en los diferentes trabajos sugieren que los cetólidos tienen un efecto antineumocócico superior al de los macrólidos y otros antimicrobianos disponibles. La actividad es independiente de su sensibilidad o resistencia a la penicilina. Mantienen una alta actividad frente a S. pneumoniae resistente a los macrólidos, bien sea por el mecanismo de la alteración de la diana mediante la producción de metilasa (MLSB) o bien por expulsión activa, si bien se ven más afectados por el primero (3, 4).
Streptococcus pyogenes
Mantienen una alta actividad frente al estreptococo beta hemolítico del grupo A, mostrando unas excelentes cifras de CMI en las cepas sensibles a los macrólidos. Estas cifras aumentan en las cepas resistentes mediante el mecanismo de expulsión activa y son más altas en las productoras de metilasa, encontrándose aislamientos resistentes a los cetólidos (aquellos S. pyogenes que expresan el gen ermB de forma constitutiva) (5).
Staphylococcus aureus
Los cetólidos muestran actividad in vitro muy buena frente a S. aureus sensible a los macrólidos, y aunque mantienen su actividad frente a las cepas que expresan la resistencia MLSB de tipo inducible, son inactivos frente a aquellos aislamientos con resistencia MLSB constitutiva.
Otros microorganismos grampositivos
Los cetólidos presentan una buena actividad frente a los enterococos sensibles a los macrólidos, si bien no son efectivos frente a los enterococos resistentes a éstos.
Frente a los estreptococos del grupo viridans (S. mitis, S. oralis, S. sanguis, etc.), este grupo de antimicrobianos muestra una excelente actividad, independientemente del comportamiento de las cepas frente a los macrólidos clásicos (6).
Los cetólidos también poseen una excelente actividad in vitro frente a especies de los géneros Lactobacillus, Pediococcus y Leuconostoc, siendo hasta diez veces más activos que los macrólidos clásicos y muy superiores a otros antimicrobianos, como los betalactámicos y las quinolonas.
En resumen, los cétolidos presentan una potencia antimicrobiana in vitro superior a la de los macrólidos (en ocasiones más de cuatro veces) frente a los microorganismos grampositivos más comúnmente aislados en infecciones respiratorias, sensibles a macrólidos, y mantienen una gran actividad frente un importante número de patógenos respiratorios grampositivos resistentes a los macrólidos.
Microorganismos gramnegativos
Frente a Haemophilus influenzae y Moraxella catarrhalis los cetólidos exhiben una notable actividad in vitro, similar a la de azitromicina, el macrólido más efectivo frente a estos patógenos (7).
Otros microorganismos
Los microorganismos intracelulares relacionados con la producción de neumonías, como Legionella pneumophila, Chlamydia pneumoniae y Mycoplasma pneumoniae, también son inhibidos por los cetólidos, mostrando actividad similar a la de los macrólidos, a excepción de frente a Mycoplasma pneumoniae, que se inhiben con una CMI más baja con los cetólidos.
Microorganismos anaerobios
Los trabajos realizados in vitro estudiando la actividad de los cetólidos frente a anaerobios han revelado resultados discrepantes según el microorganismo en cuestión, ya que se han encontrado buenos resultados frente a Propionibacterium spp., Peptostreptococcus spp., PorphyromonasPrevotella spp. y algunas especies de Clostridium como C. perfringens; sin embargo, su acción es reducida en Clostridium difficile y Bacteroides grupo fragilis. spp.,
Otro aspecto importante a considerar, además de su actividad in vitro, es la capacidad bactericida de este grupo de antimicrobianos en comparación con los macrólidos clásicos, que son primariamente bacteriostáticos. Se han realizado estudios de actividad bactericida con los patógenos más comúnmente aislados, como S. pneumoniae, S. pyogenes, H. influenzae y M. catarrhalis. Estos estudios demuestran que los resultados dependen de las concentraciones y de las cepas estudiadas. Así, frente a S. pneumoniae se han encontrado buenos resultados bactericidas a las 12 y 24 horas a concentraciones de dos veces la CMI (8), y esta cinética es más temprana a concentraciones que superan diez veces la CMI. Se han encontrado similares resultados con S. pyogenes, aunque con una cinética bactericida más lenta y dependiendo del mecanismo de resistencia presente en la cepa. Los ensayos realizados con H. influenzae y M. catarrhalis muestran que los cetólidos, a concentraciones que doblan la CMI, poseen una cinética lenta, siendo ésta más rápida cuando las concentraciones probadas son cuatro veces la CMI (7).
Por otro lado, distintos trabajos han demostrado un significativo efecto postantibiótico frente a la mayoría de los patógenos respiratorios. Estos resultados están igualmente en dependencia de la concentración utilizada, pero puede alcanzar las 8 horas en S. pneumoniae sensible o resistente a los macrólidos y en H. influenzae y M. catarrhalis productores o no de betalactamasas (9).
Por último, es imprescindible señalar que además de las propiedades antimicrobianas en sí, todos los agentes antiinfecciosos deben poseer unas características farmacocinéticas idóneas para su uso en la práctica clínica, con unos parámetros adecuados en posología, concentración máxima, área bajo la curva, difusión a tejidos y penetración intracelular (10). Los cetólidos poseen unas propiedades farmacocinéticas adecuadas que coadyuvan tanto a la curación del proceso infeccioso como al cumplimiento del tratamiento.
En resumen, la excelente actividad in vitro, la baja tasa de resistencias y su idónea farmacocinética hacen de los cetólidos una alternativa eficaz en el tratamiento de las infecciones de vías respiratorias.
Bibliografia
1. Agouridas, C., Benedetti, Y., Bonnefoy, A. y cols. Ketolides: A new class of macrolide antibacterials – Structural characteristics and biological properties of RU 6004. Curr Infect Dis Ther 1997; 21: 279-295.
2. Douthwaite, S. Structure-activity relationships of ketolides vs. macrolides. Clin Microbiol Infect 2001; 7 (Suppl. 3): 11-17.
3. Morosini, M.I., Cantón, R., Loza, E. y cols. In vitro activity of telithromycin against Spanish Streptococcus pneumoniae isolates with characterized macrolide resistance mechanisms. Antimicrob Agents Chemother 2001; 45: 2427-2431.
4. Davies, T.A., Ednie, L.M., Hoellman, D.M., Pankuch, G.A., Jacobs, M.R., Appelbaum, P.C. Antipneumococcal activity of ABT-773 compared to those of 10 other agents. Antimicrob Agents Chemother 2000; 44: 1894-1899.
5. Giovanetti, E., Montanari, M.P., Marchetti, F., Varaldo, P.E. In vitro activity of ketolides telithromycin and HMR 3004 against Italian isolates of Streptococcus pyogenes and Streptococcus pneumoniae with different erythromycin susceptibility. J Antimicrob Chemother 2000; 46: 905-908.
6. Alcaide, F., Benítez, M.A., Carratalá, J., Gudiol, F., Liñares, J., Martín, R. In vitro activities of the new ketolide HMR 3647 (telithromycin) in comparison with those of eight other antibiotics against viridans group streptococci isolated from blood of neutropenic patients with cancer. Antimicrob Agents Chemother 2001; 45: 624-626.
7. Credito, K.L., Lin, G., Pankuch, G.A., Bajaksouzian, S., Jacobs, M.R., Appelbaum, P.C. Susceptibilities of Haemophilus influenzae and Moraxella catarrhalis to ABT-773 compared to their susceptibilities to 11 other agents. Antimicrob Agents Chemother 2001; 45: 67-72.
8. Boswell, F.J., Andrews, J.M., Wise, R. Pharmacodynamic properties of HMR 3647, a novel ketolide, on respiratory pathogens, enterococci and Bacteroides fragilis demonstrated by studies of time-kill kinetics and postantibiotic effect. J Antimicrob Chemother 1998; 41: 149-153.
9. Odenholt, I., Lowdin, E., Cars, O. Pharmacodynamics of telithromycin in vitro against respiratory tract pathogens. Antimicrob Agents Chemother 2001; 45: 23-29.
10. Namour, F., Wessels, D.H., Pascual, M.H., Reynolds, D., Sultan, E., Lenfant, B. Pharmacokinetics of the new ketolide telithromycin (HMR 3647) administered in ascending single and multiple doses. Antimicrob Agents Chemother 2001; 45: 170-175.
Aportaciones microbiológicas de los cetólidos
D. Domingo
Servicio de Microbiología, Hospital Universitario de La Princesa, Madrid
El incremento de la resistencia a los macrólidos entre los patógenos respiratorios, especialmente Streptococcus pneumoniae, ha motivado la necesidad de disponer de nuevos agentes antimicrobianos que también sean eficaces frente a los aislamientos resistentes a los antimicrobianos del grupo MLSBin vitro e in vivo frente a patógenos respiratorios como S. pneumoniae, Haemophilus influenzae, Moraxella catarrhalis y Streptococcus pyogenes, muchos de los cuales son resistentes a los betalactámicos y los macrólidos. La actividad antimicrobiana de los cetólidos se extiende a patógenos respiratorios atípicos como Mycoplasma pneumoniae, Legionella spp. y Chlamydia spp. (2). (macrólidos, lincosamidas y estreptograminas del grupo B) y presenten un menor potencial para seleccionar o inducir resistencias. Un producto de este esfuerzo son los cetólidos, que son la última generación de antimicrobianos derivados del anillo macrolactona de 14 átomos (1). Los cetólidos tienen una potente actividad
La característica principal que define a los cetólidos es el cambio del azúcar cladinosa en el carbono 3 por un grupo ceto (de ahí el nombre genérico del grupo). Esto se ha conseguido reemplazando el grupo cladinosa de la eritromicina, dejando un grupo 3-hidroxilo, que es posteriormente oxidado para formar un derivado ceto; además, se añade un grupo metilo en la posición C6 (como en la claritromicina) para mejorar la estabilidad a pH ácido y prevenir el proceso de hemicetalización interna. Se creía que el azúcar l-cladinosa era necesario para la actividad antimicrobiana de los macrólidos de 14 átomos, y de hecho, en su ausencia y sin otras modificaciones, la retirada de este azúcar de la molécula de claritromicina, azitromicina y roxitromicina da lugar a la pérdida de actividad antimicrobiana. Sin embargo, la pérdida de la cladinosa puede ser compensada de forma adecuada por las modificaciones en otras posiciones del anillo macrolactónico. Éste es el caso de los cetólidos telitromicina (1) y ABT-773 (3), ambos con un grupo carbamato en C11/C12 del anillo macrolactona. La telitromicina (anteriormente HMR 3647), recientemente aprobada en Europa, y el HMR 3004 tienen una extensión alquil-aril en el grupo carbamato. El cetólido ABT-773, actualmente en fase de ensayo clínico, tiene una extensión semejante localizada en la posición 6 del anillo. A pesar de las diferencias en las posiciones de las extensiones en el anillo lactónico, los cetólidos interaccionan con sus dianas en el ribosoma bacteriano de forma similar (4). En contraste con los macrólidos de 14 y 15 átomos, los cetólidos no inducen resistencia MLSB, y esta propiedad está ligada a la ausencia del azúcar l-cladinosa.
Mecanismo De Acción
Los cetólidos y los antimicrobianos del grupo MLSB ejercen su acción antimicrobiana impidiendo la translación del RNA mensajero (RNAm) para sintetizar proteínas (5). Los ribosomas están compuestos de dos subunidades, 30S y 50S, que constan a su vez de RNA ribosómico (RNAr) y proteínas ribosómicas. La subunidad ribosómica pequeña o 30S interacciona con el RNAm y, en conjunto con el RNA de transferencia (RNAt), descodifica la información genética del RNAm. Cada vez que la subunidad 30S reconoce que un RNAt se ajusta de forma correcta con un codón RNAm, el aminoácido unido al RNAt se coloca y encaja en el lugar catalítico, denominado centro peptidil transferasa, situado en la subunidad 50S. En el centro peptidil transferasa se forma el enlace peptídico entre el nuevo aminoácido y la cadena peptídica para formar la nueva proteína. La adición secuencial de aminoácidos forma el péptido creciente, que pasa a través de un «canal peptídico de salida» en la subunidad 50S para emerger por la parte posterior del ribosoma. Este proceso puede ser bloqueado por los antimicrobianos del grupo MLSB y los cetólidos, que interaccionan con el RNAr 23S en la porción más alta del canal peptídico de salida, cerca del centro peptidil transferasa.
Recientemente se ha descrito un segundo mecanismo de acción inhibitoria de macrólidos y cetólidos, probablemente de igual importancia. Así, la formación de ribosomas nuevos requiere el ensamblado de los RNAr con numerosas proteínas ribosómicas para formar las dos subunidades ribosómicas. Se forma una subunidad 50S activa a partir de dos moléculas de RNAr (5S y 23S) y algunas de las 34 proteínas. Los macrólidos y los cetólidos interaccionan con las subunidades 50S parcialmente ensambladas, bloqueando este proceso y permitiendo así la degradación nucleotídica de las partículas precursoras no ensambladas (6).
Champney y Tober (4) han demostrado que los cetólidos telitromicina, HMR 3004, ABT 773 y los dos derivados 2-fluorados (HMR 3787 y HMR 3562) son más activos inhibiendo la síntesis (translación y ensamblado) que los macrólidos, y que los dos derivados fluorados son más activos que la telitromicina. De los datos aportados por estos autores se deriva que el efecto inhibitorio de cada componente sobre la translación y la formación de las subunidades 50S es equivalente.
La inhibición de la formación de subunidades 50S ha sido estudiada recientemente, y en el caso de la eritromicina origina la acumulación de partículas precursoras de 50S en las células. Estas partículas se unen a la eritromicina y contienen RNAr 23S y SS, pero sólo 18 de las 34 proteínas ribosómicas que forman la subunidad 50S. Estas partículas precursoras son fácilmente degradadas por las ribonucleasas celulares. Presumiblemente, el efecto inhibitorio de la formación de subunidades ribosómicas es muy semejante para todos los cetólidos (7).
Los lugares de interacción de los fármacos con el 23RNA son probablemente los mismos para ambos mecanismos inhibitorios, aunque este aspecto debe ser investigado más específicamente. Actualmente se tienen más conocimientos sobre el mecanismo molecular de la interacción de los cetólidos sobre las subunidades completas 50S que sobre la unión con las partículas precursoras.
Tanto los macrólidos como los cetólidos se unen en la misma región del RNAr 23S; sin embargo, la naturaleza y la fuerza de la interacción es diferente, ya que los cetólidos disponen de un residuo carbamato en C11/C12 (8). Los experimentos de protección química (footprinting) realizados han servido para determinar el lugar y la fuerza relativa de las interacciones de los ribosomas bacterianos y la eritromicina, la telitromicina, el HMR 3004, el RU 66252 (una versión con cladinosa del HMR 3004) y el RU 56006 (un cetólido sin el carbamato C11/C12). Estos ensayos de protección química se basan en el principio de que, cuando se une al ribosoma, el antimicrobiano interacciona con nucleótidos específicos en el RNAr y por lo tanto impide a esos nucleótidos reaccionar con agentes químicos que modificarían el RNAr (9). Estos experimentos han demostrado que la eritromicina, la claritromicina y los cetólidos (telitromicina y HMR 3004) interaccionan con los dominios II y V del RNAr 23S, y que no hay ningún contacto con otras regiones del RNAr. Según Douthwaite y cols. (10), estos fármacos protegen de la modificación química a los residuos A2058, A2059 y G2505 localizados en el asa central del dominio V, lo cual indica que existe contacto directo entre estos nucleótidos y las moléculas del antimicrobiano.
Además, la telitromicina y el HMR 3004 protegen adicionalmente el residuo A752 en el bucle «hairpin» 35 del dominio II del RNAr 23S (8, 9). Sin embargo, la eritromicina y la claritromicina dejan al nucleótido A752 más accesible a la modificación química, probablemente debido a efectos alostéricos inducidos por la unión de estos agentes al residuo A752. Así, la posición relativa de los nucleótidos de los dominios II y V en la estructura terciaria del RNAr 23S está probablemente muy próxima, y la distancia entre ellos puede ser acortada por los grupos carmabato de C11/C12, mientras que la eritromicina y la claritromicina no parece que sean capaces de hacer contacto directo con la base A752.
Además, el HMR 3004 y la telitromicina se unen con una fuerza aproximadamente diez veces superior al ribosoma bacteriano que la eritromicina. El grupo carbamato C11/C12 y su extensión pueden desempeñar un papel en esta mejora de la unión a través del dominio II del RNAr 23S. Es más, la adición de una extensión carbamato a la claritromicina (formando el compuesto RU 66252) incrementa su afinidad de unión aproximadamente en cinco veces, mientras que la retirada de la cladinosa en ausencia de extensión carbamato en C11/C12 (la denominada RU 56006) da lugar a una reducción de unas cien veces en la afinidad de unión, con la desaparición concomitante de los efectos de protección química sobre el dominio II (11).
La interacción de los cetólidos con el bucle 35 del dominio II, así como con el asa central del dominio V de la subunidad 23S del RNAr, hace plantearse si son dos lugares distintos de unión, cada uno con una molécula de cetólido, o si una sola molécula puede interaccionar con ambas regiones de forma simultánea. Esta cuestión ha sido resuelta variando las concentraciones de telitromicina en presencia de un exceso molar de ribosomas, lo que indica que las dos regiones forman un lugar de unión para una única molécula de telitromicina. Además, se sabe que esas dos regiones del RNAr están situadas muy cercanas en la estructura terciaria debido al plegamiento. Así pues, el nucleótido A752 del dominio II y los nucleótidos A2058, A2059 y G2505 delimitan el canal peptídico de salida de la subunidad ribosómica 50S, y pueden formar un «bolsillo» dentro del cual una única molécula de antimicrobiano interaccione con ambos dominios (II y V) a la vez (10).
Garza-Ramos y cols. (12) han comunicado recientemente la existencia de otro residuo situado en el dominio V de la subunidad 23S del RNAr, el U2609, que interacciona tanto con las moléculas de cetólidos como con las de macrólidos que contienen el azúcar cladinosa, pero que lo hace de forma mucho más intensa en el caso de los cetólidos. Dicha interacción parece ser importante y contribuir en gran medida a la unión de la molécula de cetólido con el ribosoma. Así pues, actualmente se consideran como nueleótidos importantes involucrados en la unión los residuos 2057 a 2059, 2609 y 2611 en el dominio V, y el nucleótido 752 en el dominio II. La mutación del nucleótido U2609C confiere resistencia exclusivamente a los cetólidos, mientras que se incrementa la sensibilidad bacteriana a los cladinosólidos.
Todos estos nucleótidos forman una agrupación espacial localizada a una distancia aproximada de un tercio de la zona de interfase de la subunidad ribosómica 50S. El lugar de unión del fármaco está localizado cerca del fondo de una cavidad conteniendo el centro peptidil transferasa, en la entrada del túnel de formación del péptido naciente. La forma más sencilla de que el fármaco alcance dicha localización es en el momento en que la subunidad 50S está todavía aislada. Sin embargo, dado el relativo gran tamaño del túnel de salida del péptido y de su hipotética propiedad de no ser fácilmente colapsable, es también probable que el antimicrobiano difunda al lugar de acción a través del túnel.
Garza-Ramos y cols. (12) han propuesto que la protección a la modificación química de los cetólidos en los experimentos de footprinting refleja la existencia de un contacto directo entre U2609 y una molécula de cetólido (probablemente involucrando al grupo 3-ceto). Según estos autores, la protección de A752 se afecta por el grupo 3-ceto y por las extensiones aril-alquil, y la localización de estas cadenas no parece relevante, protegiendo de igual modo cuando están situadas en C11/C12 (como en la telitromicina) que cuando lo están en el carbono C6 (como en el cetólido ABT 773), pero que depende en gran medida de la naturaleza química de dicha cadena. La interacción del cetólido y el nucleótido U2609 y el grupo 3-ceto puede tener lugar de forma que la molécula de cetólido quede en posición favorable para proteger a A752 de la modificación química.
Así pues, el lugar de unión del macrólido/cetólido parece lógico y conforme con el mecanismo de acción propuesto, según el cual el fármaco bloquea la translación impidiendo estéricamente el crecimiento del péptido naciente, ya que se localiza en la parte más estrecha del canal de salida, y sólo bloquea los ribosomas que comienzan la formación del péptido, es decir, con péptidos cortos, entre dos y cinco aminoácidos, ya que cuando el péptido es más largo ya ha pasado el lugar estrecho del túnel y el fármaco es incapaz de bloquearlo (13).
Perfil Y Mecanismo De Resistencia
Los cetólidos tienen actividad frente a S. pneumoniae resistente a los macrólidos por mecanismo de flujo (genes mef), al igual que frente a aquellos aislamientos resistentes por poseer una metilasa (genes erm) de carácter inducible o constitutivo y que confiere resistencia a los antimicrobianos del grupo MLSB (14). En cuanto a S. pyogenes y Staphylococcus aureus, la telitromicina es eficaz frente a la mayoría (pero no todas) de las cepas resistentes a la eritromicina de forma constitutiva (15).
Uno de los mecanismos por los que una bacteria se puede convertir en resistente a los macrólidos es por una alteración de la estructura del centro de unión del fármaco en el ribosoma. En el caso de la resistencia por genes erm, esto se consigue mediante la expresión de una metiltransferasa, que metila la base A2058 y confiere resistencia MLSB. También puede aparecer este fenotipo por mutaciones en los nucleótidos importantes en la unión del fármaco con la subunidad 23S del RNAr ribosómico (16). Recientemente se han descrito mutaciones en las proteínas ribosómicas LA y L22, cercanas al lugar de acción de los antimicrobianos, es decir, adyacentes a los nucleótidos A2058 y A752, respectivamente, que confieren resistencia por alterar la conformación del lugar de unión del fármaco (17).
Los cetólidos con un residuo carbamato permanecen activos frente a la mayoría de los aislamientos resistentes a la eritromicina, probablemente a causa de las diferencias en el contacto del fármaco con el núcleo de acción. Esta idea también viene apoyada por los experimentos de footprinting sobre ribosomas con mutación en A2058, pasando a G2058. Esta mutación reduce la unión de la eritromicina y de la claritromicina a los ribosomas bacterianos aproximadamente 10.000 veces; sin embargo, la unión de los cetólidos es sustancialmente reducida, pero se mantiene unas 20 veces superior a la de la eritromicina.
La resistencia bacteriana a los cetólidos es poco frecuente y de reciente descripción. Así, se han descrito varios mecanismos de resistencia: a) cambios en algunas de las bases de la subunidad 23S del RNAr implicadas en la unión del cetólido con el ribosoma; b) mutaciones en la secuencia de la proteína L4; y c) minipéptidos.
Mutaciones en las bases del RNAr 23S implicadas en la unión
Se ha comprobado que la telitromicina selecciona mutantes resistentes con menos frecuencia que la eritromicina, la claritromicina, la azitromicina, la roxitromicina, la clindamicina y la pristinamicina tras 50 pases de cinco aislamientos de S. pneumoniae sensibles a los macrólidos y seis resistentes conteniendo los genes mefE o ermB, a concentraciones subinhibitorias de eritromicina (18). Sólo aparecieron tres mutantes resistentes a la telitromicina (CMI >1 mg/l), los tres con CMI <8 mg/l.
Tait-Kamradt y cols. (19, 20) estudiaron el mecanismo de resistencia de varias mutantes resistentes a los macrólidos y los cetólidos, seleccionadas tras varios pases en presencia de azitromicina. Ninguna de las mutaciones en C2611A, A2058G y A5059G confirió resistencia de alto grado a la telitromicina. Sin embargo, el cambio A2611G aumentó 130 veces la CMI de telitromicina. También encontraron cambios en la secuencia conservada 63 a 74 de la proteína ribosómica L4, sin que afectaran a la sensibilidad a la telitromicina.
Garza-Ramos y cols. (12) describieron una mutación en la base 2609 (transición U2609C) que confiere resistencia a los cetólidos, pero incrementa ligeramente la sensibilidad a los derivados que contienen cladinosa (incluso el derivado de la telitromicina con cladinosa). De estos estudios se desprende la importancia de la unión de los cetólidos a dicho residuo.
Cambios en la secuencia de la proteína ribosómica L4
Tait-Kamradt y cols. (19, 20) estudian los mecanismos de resistencia de 20 aislamientos clínicos de S. pneumoniae con fenotipos extraños o poco frecuentes (como el ML y el MSB, que no son portadores de genes ermB o mefA), y detectan un único aislamiento MSB procedente de Canadá con una inserción de seis aminoácidos en la proteína L4 (69GTGREKGTGRAR71), con impacto sobre su crecimiento (mejor a 35 ºC) y que incrementa en 500 veces la CMI de la telitromicina.
Resistencia por minipéptidos o péptidos cortos
Tripathi y cols. (13) describieron en 1998 un nuevo mecanismo que confiere resistencia a los cetólidos y los macrólidos, a causa de unos péptidos de resistencia codificados por regiones localizadas en la subunidad 23S del RNAr, normalmente «escondido» de la maquinaria de translación de la célula bacteriana. La expresión de esos genes puede ocurrir por mutación o degradación del RNAr o por activación de esos minigenes silentes en el ribosoma. La translación ocurre en los ribosomas, y los péptidos cortos que emergen «expulsan» al antimicrobiano del ribosoma (mecanismo de clarificación ribosomal, también denominado de bottlebrush o de «cepillo de botella»). Existe una correlación específica entre la secuencia de estos minipéptidos y la estructura del antimicrobiano de que se trate, sin detectarse resistencia cruzada con otras familias de antimicrobianos. Los minipéptidos interaccionan con la estructura molecular del fármaco específicamente, tanto que se han descrito secuencias distintas entre los que confieren resistencia a la eritromicina, la claritromicina y la telitromicina.
Así pues, los cetólidos son un nuevo grupo de antimicrobianos derivados del anillo macrolactona de la eritromicina, que mejoran su actividad e incluso la de sus derivados más recientes. No inducen resistencia MLSB en las cepas portadoras de genes erm y mejoran la interacción con el ribosoma. La interacción con los dominios II y V de la subunidad 23S del RNAr incrementa la capacidad del antimicrobiano para inhibir el ensamblado de nuevas subunidades ribosómicas 50S y para bloquear la síntesis de proteínas sobre las unidades ribosómicas ya formadas. Estas propiedades hacen que los cetólidos sean fármacos con una potente actividad antimicrobiana frente a un amplio espectro de patógenos respiratorios.
Bibliografía
1. Bryskier, A. New research in macrolides and ketolides since 1997. Expert Opin Invest Drugs 1999; 8: 1171-1194.
2. Balfour, J.A., Figgitt, D.P. Telithromycin. Drugs 2001; 61: 815-829.
3. Barrett, J.F., Dougherty, T.J. ABT-773: A new ketolide antibiotic. Expert Opin Invest Drugs 2001; 10: 343-351.
4. Champney, W.S., Tober, C.L. Structure-activity relationships for six ketolide antibiotics. Current Microbiol 2001; 42: 201-210.
5. Capobianco, J.O., Cao, Z.S., Shortidge, V.D., Ma, Z., Flamm, R.K., Zhong, P. Studies of the novel ketolide ABT-773: Transport, binding to ribosomes, and inhibition of protein synthesis in Streptococcus pneumoniae. Antimicrob Agents Chemother 2000; 44: 1562-1567.
6. Champney, W.S. Bacterial ribosomal subunit synthesis: A novel antibiotic target. Current Drug Targets Infectious Disorders 2001; 1: 19-36.
7. Usary, J., Cjampney, W.S. Erytrhomycin inhibition of 50S ribosomes subunit formation in Escherichia coli cells. Mol Microbiol 2001; 40: 951-962.
8. Hansen, L.H., Mauvais, P., Douthwaite, S. The macrolide-ketolide antibiotic binding site is formed by structures in domains II and V of 23S ribosomal RNA. Mol Microbiol 1999; 31: 623-631.
9. Xiong, L., Shah, S., Mauvais, P., Mankin, A.S. A ketolide resistance mutation in domain II of 23S rRNA reveals the proximity of hairpin 35 to the peptidyl transferase centre. Mol Microbiol 1999; 31: 633-639.
10. Douthwaite, S., Hansen, L.H., Mauvais, P. Macrolide-ketolide inhibition of MLS resistant ribosomes is improved by alternative drug interaction with domain II of 23S rRNA. Mol Microbiol 2000; 36: 183-193.
11. Douthwaite, S., Champney, W.S. Structures of ketolides and macrolides determine their mode of interaction with the ribosomal target site. J Antimicrob Chemother 2001; 48 (Suppl. B): 1-8.
12. Garza-Ramos, G., Xiong, L., Zhong, P., Mankin, A. Binding site of macrolide antibiotics on the ribosome: New resistance mutation identifies a specific interaction of ketolides with rRNA. J Bacteriol 2001; 183: 6898-6907.
13. Tripathi, S., Kloss, P.S., Mankin, A.S. Ketolide resistance conferred by short peptides. J Biol Chem 1998; 273: 20073-20077.
14. Pankuch, G.A., Visalli, M.A., Jacobs, M.R., Appelbaum, P.C. Susceptibilities of penicillin- and erytrhomycin-susceptible and -resistant pneumococci to HMR 3647 (RU 66647), a new ketolide, compared with susceptibilities to 17 other agents. Antimicrob Agents Chemother 1998; 42: 624-630.
15. Bemer-Melchior, P., Juvin, M.E., Tassin, S., Bryskier, A., Schito, G.C., Drugeon, H.B. In vitro activity of the new ketolide telithromycin compared with those macrolides against Streptococcus pyogenes: Influences of resistance mechanisms and methodological factors. Antimicrob Agents Chemother 2000; 44: 2999-3002.
16. Vester, B., Douthwaite, S. Macrolide resistance conferred by base substitutions in 23S rRNA. Antimicrob Agents Chemother 2001; 45: 1-12.
17. Yusupov, M.M., Yusupova, G.Z., Baucom, A., Lieberman, K., Earnest, T.N., Cate, J.H. Crystal structure of the ribosome at 5.5 A resolution. Science 2001; 292: 883-896.
18. Davies, T.A., Dewasse, B.E., Jacobs, M.R., Appelbaum, P.C. In vitro development of resistance to telithromycin (HMR 3647), four macrolides, clindamycin, and pristinamycin in Streptococcus pneumoniae. Antimicrob Agents Chemother 2000; 44: 414-417.
19. Tait-Kamradt, A., Davies, T., Appelbaum, P.C. y cols. Two new mechanisms of macrolide resistance in clinical strains of Streptococcus pneumoniae from Eastern Europe and North America. Antimicrob Agents Chemother 2000; 44: 3395-3401.
20. Tait-Kamradt, A., Davies, T., Cronan, M., Jacobs, M.R., Appelbaum, P.C., Sutcliffe, J. Mutations in 23S rRNA and ribosomal protein LA account for resistance in pneumococcal strains selected in vitro by macrolide passage. Antimicrob Agents Chemother 2000; 44: 2118-2125.
Eficacia terapéutica de los cetólidos
J. Barberán
Servicio de Enfermedades Infecciosas, Hospital Gómez Ulla; Departamento de Medicina, Universidad Complutense, Madrid
La telitromicina deriva estructuralmente de los macrólidos de 14 átomos de carbono, donde la lactona macrocíclica tiene una sustitución ceto en el carbono 3, en vez de la cladinosa, que evita la inducción de resistencias bacterianas del tipo MLSB; un grupo metoxi en la posición 6 que le da estabilidad en medio ácido; y una cadena carbamato en C11-C12, con la que obtiene una mayor afinidad por los ribosomas y por ende una mejor actividad antibacteriana (Fig. 1). Su mecanismo de acción es similar al de los macrólidos, es decir, inhiben la síntesis proteica mediante su unión a la fracción 50S de los ribosomas, pero con una afinidad seis a diez veces superior a la de la eritromicina y la claritromicina (2, 3).
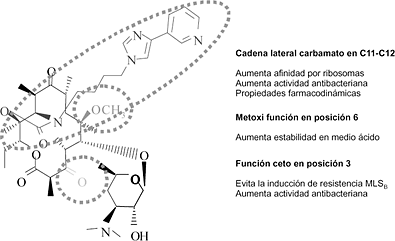
Figura 1. Estrucutra molecular de telitromicina
Los cetólidos son una nueva clase de antibióticos pertenecientes al grupo formado por los macrólidos, las lincosamidas y las estreptograminas (MLS), del cual la telitromicina es el primer y único representante disponible para uso clínico (1).
Espectro Microbriano
La telitromicina, junto con las fluoroquinolonas, son los únicos antibióticos cuyo espectro de acción incluye a los principales microorganismos causantes de las infecciones respiratorias adquiridas en la comunidad: Streptococcus pyogenes, incluido el resistente a la eritromicina, Streptococcus pneumoniae tanto sensible como resistente a la penicilina y los macrólidos, Haemophilus influenzae, Moraxella catarrhalis y agentes atípicos como Mycoplasma pneumoniae, Chlamydia pneumoniae y Legionella pneumophila. Además, la telitromicina tiene actividad frente a algunas micobacterias y anaerobios tales como Peptostreptococcus spp., Prevotella spp., Actinomyces spp., algunas Porphyromonas spp. y Clostridium perfringens, pero no sobre los bacilos gramnegativos (4) (Tabla 1).
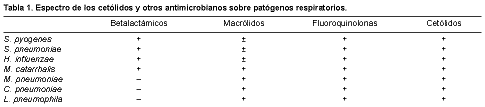
Los puntos de corte establecidos por el NCCLS para considerar sensibles a la telitromicina a los distintos microorganismos son los siguientes: para estreptococos, estafilococos y enterococos 1 mg/l, y para H. influenzae 2 mg/l. Las CMI90 obtenidas para las bacterias probadas están muy por debajo de estos puntos: para S. pyogenes (sensible y resistente a la eritromicina) 0,008-1 mg/l; para S. pneumoniae sensible a la eritromicina y la penicilina 0,008-0,06 mg/l, y para el resistente 0,06-0,5 mg/l; para H. influenzae 2 mg/l; para M. catarrhalis 0,03-0,25 mg/l; para L. pneumophilaM. pneumoniae 0,015 mg/l; y para C. pneumoniae 0,06-0,25 mg/l (4). 0,03-0,12 mg/l; para
Perfil Farmacocinético Y Farmacodinmico
La telitromicina se absorbe por vía oral, con una biodisponibilidad del 57%, que no se ve afectada por la ingestión de alimentos. Con una única dosis de 800 mg el pico sérico (Cmáx) es de aproximadamente 2 mg/l y se alcanza entre una y dos horas tras su administración, manteniéndose así durante todo el tratamiento; el área bajo la curva (ABC) supera las 7 horas, lo cual hace que los índices predictores de éxito terapéutico (Cmáx/CMI y ABC/CMI) con los que se ha relacionado sean altos. A esto hay que añadir la gran penetración en los tejidos de las vías respiratorias (senos paranasales, amígdala, mucosa bronquial y líquido epitelial), donde la concentración del antibiótico supera en varias veces a la plasmática, y muy particularmente en el macrófago alveolar, con un ratio mayor de 300, de gran importancia para el tratamiento de las neumonías atípicas. En la saliva también alcanza concentraciones superiores a la plasmática, disminuyendo el riesgo de estado de portador, tan importante en la transmisión de las infecciones respiratorias. Su vida media de más de 10 horas permite la administración en dosis única diaria, lo cual redunda en la comodidad del paciente y en el cumplimiento terapéutico. El 70% del antibiótico se metaboliza en el hígado mediante oxidación microsomal en el citocromo P-450 CYP 34A y se elimina principalmente por heces (75%) en forma de varios metabolitos inactivos (Tablas 2 a 4) (5).



Experiencia Clenida
Hasta la fecha se han realizado varios ensayos clínicos internacionales, comparativos y no comparativos, en fase III. En la faringoamigdalitis, la telitromicina, en dosis única de 800 mg/24 horas durante cinco días, se ha comparado con fenoximetilpenicilina (500 mg/8 horas) y claritromicina (500 mg/12 horas) durante diez días, obteniéndose resultados clínicos y microbiológicos similares (6, 7) (Fig. 2). En sinusitis maxilar aguda, la telitromicina (800 mg/24 horas) durante cinco días se comportó de forma similar a una pauta de diez días del mismo antibiótico y que amoxicilina-ácido clavulánico (500-125 mg/8 horas) y cefuroxima axetilo (500 mg/12 horas) también durante diez días (8, 9) (Fig. 3). En la exacerbación de la bronquitis crónica, el tratamiento de cinco días con telitromicina (800 mg/24 horas) ha tenido un efecto parecido al de diez días con amoxicilina-ácido clavulánico (500-125 mg/8 horas) y cefuroxima axetilo (500 mg/12 horas) (10, 11) (Fig. 4). En la neumonía adquirida en la comunidad, la telitromicina durante diez días también ha sido igual de eficaz que el mismo tiempo con amoxicilina (1 g/8 horas), claritromicina (500 mg/12 horas) o trovafloxacino (200 mg/24 horas) (12-14) (Fig. 5).
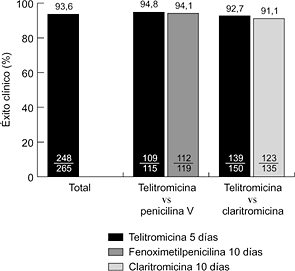
Figura 2. Eficacia clínica de telitromicina en faringoamigdalitis (6, 7).
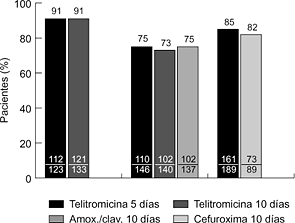
Figura 3. Eficacia clínica de telitromicina en sinusitis maxilar aguda (8, 9).
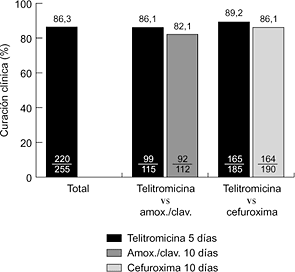
Figura 4. Eficacia clínica de telitromicina en exacerbación de la EPOC (10, 11).
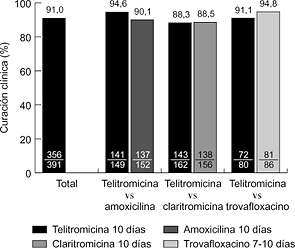
Figura 5. Eficacia clínica de telitromicina en neumonía comunitaria (12-14).
Tolerabilidad
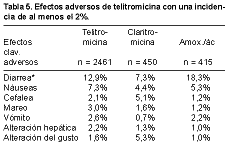
Los efectos adversos de tipo gastrointestinal son los más frecuentes, en particular la diarrea (12,9%) y las náuseas (7,3%). La primera ha sido significativamente mayor que en los comparadores, excepto con amoxicilina-ácido clavulánico. No se han observado prolongaciones de consideración del intervalo QT en el electrocardiograma (4, 15) (Tabla 5).
En cuanto a las interacciones medicamentosas, se han apreciado aumentos de la concentración plasmática y del área bajo la curva de varias veces con fármacos que inhiben el citocromo P-450 CYP3A4, como el itraconazol y el ketoconazol, y con otros que utilizan esta misma vía microsomal, como cisaprida, simvastatina y midazolam (15).
La eliminación es fundamentalmente fecal y parece que no es necesario ajustar la dosis en los pacientes con insuficiencia renal o hepática, ni en los ancianos (4, 15).
Conclusiones
La telitromicina parece constituir una alternativa de primera línea en el tratamiento de las infecciones respiratorias adquiridas en la comunidad (faringoamigdalitis, sinusitis aguda, exacerbación de la bronquitis crónica y neumonía), debido a su actividad frente a la mayoría de los patógenos causantes de estos procesos, incluidos los resistentes a la penicilina y los macrólidos. Su perfil farmacocinético permite una dosis diaria y facilita el cumplimiento terapéutico, y su tolerabilidad es buena.
Los ensayos clínicos que en estos momentos se están realizando y la vigilancia postcomercialización nos darán la verdadera dimensión de la telitromicina en la infección respiratoria comunitaria, que sin duda alguna parece destinada a ocupar un lugar prioritario.
Bibliografía
1. Bryskier, A., Agouridas, C., Chantot, J. Ketolide: Novel antibacterial agent designed to overcome erythromycin A resistance. En: Zinner, S.H., Young, L.S., Acar, J.F. y cols. (Eds.). New considerations for macrolides, azalides, streptogramins and ketolides. Marcel-Dekker, New York 2000; 79-102.
2. Hansen, L.H., Mauvais, P., Douthwaite, S. The macrolide-ketolide antibiotic binding site is formed by structures in domains II and V of 23S ribosomal RNA. Mol Microbiol 1999; 31: 623-631.
3. Douthwaite, S., Hansen, L.H., Mauvais, P. Macrolide-ketolide inhibition of MLS-resistant ribosomes is improved by alternative drug interaction with domain II of 23S rRNA. Mol Microbiol 2000; 36: 183-193.
4. Bearden, D.T., Neuhauser, M.M., Garey, K.W. Telithromycin: An oral ketolide for respiratory infections. Pharmacotherapy 2001; 21: 1204-1222.
5. Namour, F., Wessels, D., Pascual, M. y cols. Pharmacokinetics of the new ketolide telithromycin (HMR 3657) administered in ascending single and multiple doses. Antimicrob Agents Chemother 2001; 45: 170-175.
6. Norrby, S., Bacart, P., Rabie, W. y cols. Efficacy of 5 days telithromycin (HMR 3647) vs. 10 days penicillin V in the treatment of pharyngitis in adults. 40th Interscience Conference on Antimicrobial Agents and Chemotherapy. American Society for Microbiology, Washington, DC 2000; 489.
7. Ziter, P., Quinn, J., Leroy, B. y cols. Oral telithromycin (HMR 3647; 800 mg OD) for 5 days is well tolerated and as effective as clarithromycin (250 mg BID) for 10 days in group A b-hemolytic streptococcal pharyngitis/tonsillitis. 40th Interscience Conference on Antimicrobial Agents and Chemotherapy. American Society for Microbiology, Washington, DC 2000; 472.
8. Tellier, G., Lasko, B., Leroy, B. y cols. Oral telithromycin (HMR 3647; 800 mg OD) for 5 days and 10 days is well tolerated and as effective as amoxicillin/clavulanic acid (500/125 mg TID) for 10 days in acute maxillary sinusitis (AMS) in adults. 40th Interscience Conference on Antimicrobial Agents and Chemotherapy. American Society for Microbiology, Washington, DC 2000; 471.
9. Roos, K., Brunswig-Pitschner, C., Kostrica, R. y cols. Efficacy and tolerability of a 5-day course of a new ketolide antimicrobial, telithromycin (HMR 3647), for the treatment of acute sinusitis. 40th Interscience Conference on Antimicrobial Agents and Chemotherapy. American Society for Microbiology, Washington, DC 2000; 490.
10. Aubier, M., Aldons, P., Leak, A. y cols. Efficacy and tolerability of a 5-day course of a new ketolide antimicrobial, telithromycin (HMR 3647), for the treatment of acute exacerbations of chronic bronchitis (AECB) in patients with COPD. 40th Interscience Conference on Antimicrobial Agents and Chemotherapy. American Society for Microbiology, Washington, DC 2000; 489.
11. DeAbate, C., Heyder, A., Leroy, B. y cols. Oral telithromycin (HMR3647; 800 mg od) for 5 days is well tolerated and as effective as cefuroxime axetil (500 mg bid) for 10 days in adults with acute exacerbations of chronic bronchitis (AECB). 40th Interscience Conference on Antimicrobial Agents and Chemotherapy. American Society for Microbiology, Washington, DC 2000; 471.
12. Hagberg, L., Torres, A., Van Rensburg, D. y cols. Efficacy and tolerability of telithromycin (HMR 3647) vs. high-dose amoxicillin in the treatment of community-acquired pneumonia. 40th Interscience Conference of Antimicrobial Agents and Chemotherapy. American Society for Microbiology, Washington, DC 2000; 490.
13. Tellier, G., Hassman, J., Leroy, B. y cols. Oral telithromycin (HMR3647; 800 mg od) is well tolerated and as effective as oral clarithromycin (500 mg bid) in community-acquired pneumonia (CAP) in adults. 40th Interscience Conference of Antimicrobial Agents and Chemotherapy. American Society for Microbiology, Washington, DC 2000; 471.
14. Pullman, J., Champlin, J., Leroy, B. y cols. Oral telithromycin (HMR 3647; 800 mg OD) for 7-10 days is well tolerated and as effective as oral trovafloxacin (200 mg OD) for 7-10 days in community-acquired pneumonia (CAP) in adults. 40th Interscience Conference on Antimicrobial Agents and Chemotherapy. American Society for Microbiology, Washington, DC 2000; 472.
15. Barman, J.A., Figgitt, D.P. Telithromicyn. Drugs 2001; 61: 815-829.
Conducta terapéutica ante infecciones hospitalarias y de la comunidad por grampositivos
J.M. Aguado
Hospital Universitario 12 de Octubre, Madrid
En las presentaciones anteriores se ha comentado en extenso el incremento que están sufriendo las infecciones por grampositivos. En los pacientes neutropénicos, por ejemplo, nos estamos acostumbrando a ver cada vez con más frecuencia pacientes con bacteriemias por microorganismos como Streptococcus viridans, o por otros microorganismos grampositivos menos frecuentes como Corynebacterium jeikeium o Bacillus spp. A continuación comentaremos cuál está siendo la evolución y la tendencia de la etiología y la clínica de las infecciones por grampositivos en el hospital y en la comunidad, de qué nuevas alternativas terapéuticas disponemos frente a ellas y qué conducta terapéutica hemos de seguir.
Los tres patógenos que centran nuestra atención son Streptococcus pneumoniae, como principal agente de infección respiratoria en la comunidad, y Staphylococcus aureus resistente a meticilina (SARM) y Enterococcus resistente a vancomicina como agentes productores de infección hospitalaria. El problema presentado por la resistencia a los antibióticos de estas bacterias es cada vez más importante y grave, ya que se asocia con incrementos de la mortalidad que, por ejemplo, son evidentes en el caso de la bacteriemia por SARM, condicionando una mortalidad cuatro veces mayor que la de los pacientes cuya infección es por S. aureus sensible a la meticilina, o en la neumonía por SARM, en cuyo caso la mortalidad esperable es 20 veces mayor que si S. aureus es sensible a la meticilina. Para S. pneumoniae no se ha podido demostrar todavía un incremento de la mortalidad en las neumonías causadas por cepas con elevada resistencia a la penicilina, quizá debido al escaso número de casos descritos con CMI muy elevadas de este antibiótico. Sin embargo, se sabe que las cepas de neumococo altamente resistentes a la penicilina producen fracaso terapéutico en los pacientes con meningitis neumocócica con mucha más frecuencia que si la meningitis es causada por una cepa sensible.
Es evidente que la resistencia plantea problemas terapéuticos. En el caso de SARM, el antibiótico usado hasta este momento ha sido mayoritariamente la vancomicina. Conviene recordar, no obstante, que este antibiótico tiene indudables defectos que no lo hacen ideal. En primer lugar, su lenta actividad bactericida condiciona a veces la persistencia de una bacteriemia a pesar de alcanzar concentraciones séricas adecuadas y con CMI dentro del rango de sensibilidad. Por otro lado, en los últimos años se ha demostrado resistencia o sensibilidad intermedia in vitro de cepas de S. aureus y S. epidermidis a los glucopéptidos (GISA), y tolerancia de algunas cepas de S. pneumoniae a la vancomicina. Por último, la vancomicina tiene una serie de efectos secundarios bien conocidos, como la nefrotoxicidad o las reacciones cutáneas.
Existen nuevas opciones terapéuticas para las infecciones por cocos grampositivos multirresistentes. En las infecciones hospitalarias por SARM y enterococos resistentes a los glucopéptidos, los dos antibióticos de que disponemos son la quinupristina-dalfopristina y el linezolid.
La quinupristina-dalfopristina es una alternativa al tratamiento con glucopéptidos. El espectro de este nuevo antibiótico se extiende a cocos grampositivos incluyendo cepas resistentes de Enterococcus faecium, pero no de Enterococcus faecalis, bacilos grampositivos y algunos patógenos respiratorios intracelulares. Ciertos efectos adversos, como la aparición de frecuentes flebitis si se infunde a través de una vía venosa periférica, dificultan su uso, si bien este problema puede ser superado administrando el fármaco por una vía central.
El linezolid constituye también una nueva alternativa a los glucopéptidos. Sería de elección en el tratamiento de las infecciones leves a moderadas por estos microorganismos, ya que puede administrarse por vía oral (con una biodisponibilidad similar a la de la vía intravenosa), pero también permitiría la terapia secuencial en caso de infecciones más graves, lo que lo convierte en una gran expectativa terapéutica en la infección relacionada con catéter venoso. Es activo frente a cocos grampositivos, incluyendo cepas resistentes (SARM, neumococo resistente a penicilina, enterococo resistente a vancomicina), bacilos grampositivos y algunos patógenos respiratorios, así como algunos anaerobios como Peptostreptococcus, Prevotella, Fusobacterium nucleatum). Está indicado en el tratamiento de la neumonía adquirida en la comunidad, la neumonía nosocomial y la infección de piel y partes blandas. El efecto adverso más preocupante de este antibiótico es su potencial capacidad mielosupresora, si bien este efecto parece reversible una vez retirado el fármaco.
En la infección comunitaria también contamos con nuevas opciones terapéuticas frente a la infección por grampositivos, fundamentalmente con quinolonas (levofloxacino y moxifloxacino) y telitromicina. Este último antibiótico es una excelente alternativa a los macrólidos en la infección respiratoria.
En este terreno de la infección respiratoria por grampositivos se plantea la duda de si elegir antibióticos refrendados por su largo uso, como son los betalactámicos y los macrólidos, o bien los nuevos antibióticos (quinolonas o telitromicina) cuya eficacia in vitro es superior a la de los previos, pero con los que existe una menor experiencia. En este sentido, cada clínico deberá elegir según las consideraciones particulares de cada caso. Existe un miedo a que el empleo indiscriminado de estos nuevos antibióticos, especialmente de fluoroquinolonas, dispare la resistencia de los grampositivos a estos antibióticos. Hay que decir, sin embargo, que a pesar de estar en uso desde hace ya varios años, la resistencia a las quinolonas en los grampositivos no se ha incrementado o lo ha hecho muy levemente si lo comparamos con lo que ha sucedido con los macrólidos o los betalactámicos, y con lo que ocurre con la resistencia a las quinolonas de los gramnegativos, que en España ha aumentado sustancialmente. Tanto las nuevas quinolonas como la telitromicina tienen un espectro tan amplio que son fármacos difícilmente mejorables para el tratamiento de la infección respiratoria adquirida en la comunidad.
Bibliografía
– Cercenado, E., Sánchez-Carrillo, C., Alcalá, L., Bouza, E. Current status of resistance of Staphylococcus in Spain. 4th National Study, 1996. Work Group on the Study of Staphylococcus. Rev Clin Esp 1997; 197 (Suppl. 2): 12-18.
– Cisneros, J.M., Muñoz, P., Torre-Cisneros, J. y cols. Pneumonia after heart transplantation: A multi-institutional study. Spanish Transplantation Infection Study Group. Clin Infect Dis 1998; 27: 324-331.
– Conterno, L.O., Wey, S.B., Castelo, A. Risk factors for mortality in Staphylococcus aureus bacteremia. Infect Control Hosp Epidemiol 1998; 19: 32-37.
– Cordero, E., Pachón, J., Rivero, A. y cols. Community-acquired bacterial pneumonia in human immunodeficiency virus-infected patients: Validation of severity criteria. The Grupo Andaluz para el Estudio de las Enfermedades Infecciosas. Am J Respir Crit Care Med 2000; 162: 2063-2068.
– Drew, R.H., Perfect, J.R., Srinath, L., Kurkimilis, E., Dowzicky, M., Talbot, G.H. Treatment of methicillin-resistant Staphylococcus aureus infections with quinupristin-dalfopristin in patients intolerant of or failing prior therapy. Synercid Emergency-Use Study Group. J Antimicrob Chemother 2000; 46: 775-784.
– Fridkin, S.K. Vancomycin-intermediate and -resistant Staphylococcus aureus: What the infectious disease specialist needs to know. Clin Infect Dis 2001; 32: 108-115.
– Kaplan, S.L., Mason, E.O. Jr. Management of infections due to antibiotic-resistant Streptococcus pneumoniae. Clin Microbiol Rev 1998; 11: 628-644.
– Linden, P.K., Pasculle, A.W., Máñez, R. y cols. Differences in outcomes for patients with bacteremia due to vancomycin-resistant Enterococcus faecium or vancomycin-susceptible E. faecium. Clin Infect Dis 1996; 22: 663-670.
– Martone, W.J. Spread of vancomycin-resistant enterococci: Why did it happen in the United States? Infect Control Hosp Epidemiol 1998; 19: 539-549.
– McDonald, C.J. Is the placebo powerless? N Engl J Med 2001; 345: 1276-1277.
– Moellering, R.C., Linden, P.K., Reinhardt, J.,Blumberg, E.A., Bompart, F., Talbot, G.H. The efficacy and safety of quinupristin/dalfopristin for the treatment of infections caused by vancomycin-resistant Enterococcus faecium. Synercid Emergency-Use Study Group. J Antimicrob Chemother 1999; 44: 251-261.
– Pachón, J., Prados, M.D., Capote, F., Cuello, J.A., Garnacho, J., Verano, A. Severe community-acquired pneumonia. Etiology, prognosis and treatment. Am Rev Respir Dis 1990; 142: 369-373.
– Pallarés, R., Linares, J., Vadillo, M. y cols. Resistance to penicillin and cephalosporin and mortality from severe pneumococcal pneumonia in Barcelona, Spain. N Engl J Med 1995; 333: 474-480.
– Quagliarello, V.J., Scheld, W.M. Treatment of bacterial meningitis. N Engl J Med 1997; 336: 708-716.
– Rello, J., Torres, A., Ricart, M. y cols. Ventilator-associated pneumonia by Staphylococcus aureus. Comparison of methicillin-resistant and methicillin-sensitive episodes. Am J Respir Crit Care Med 1994; 150: 1545-1549.
– Rubinstein, E., Prokocimer, P., Talbot, G.H. Safety and tolerability of quinupristin/dalfopristin: Administration guidelines. J Antimicrob Chemother 1999; 44 (Suppl. A): 37-46.
– Suppola, J.P., Volin, L., Valtonen, V.V., Vaara, M. Overgrowth of Enterococcus faecium in the feces of patients with hematologic malignancies. Clin Infect Dis 1996; 23: 694-697.
– Torres, C., Reguera, J.A., Sanmartín, M.J., Pérez-Díaz, J.C., Baquero, F. vanA-mediated vancomycin-resistant Enterococcus spp. in sewage. J Antimicrob Chemother 1994; 33: 553-561.
– Waterer, G.W., Somes, G.W., Wunderink, R.G. Monotherapy may be suboptimal for severe bacteremic pneumococcal pneumonia. Arch Intern Med 2001; 161: 1837-1842.